Effects of Mood Stabilizers: Implications for the
Pathophysiology and Treatment of
Manic–Depressive Illness
Husseini K. Manji, Gregory J. Moore, and Guang Chen
Recent neuroimaging studies have demonstrated regional central nervous system volume reductions in mood disor-ders, findings that are complemented by postmortem observations of cell atrophy and loss. It is thus noteworthy that lithium and valproate have recently been demon-strated to robustly increase the expression of the cytopro-tective protein bcl-2 in the central nervous system. Chronic lithium not only exerts neuroprotective effects in several preclinical paradigms but also enhances hip-pocampal neurogenesis. Valproate robustly promotes neu-rite outgrowth and activates the ERK mitogen-activated protein kinase pathway, a signaling pathway utilized by many endogenous neurotrophic factors. Consistent with its preclinical neurotrophic/neuroprotective effects, chronic lithium treatment of patients with manic– depressive ill-ness increases brain N-acetylaspartate (a putative marker of neuronal viability and function) levels, an effect that is localized almost exclusively to gray matter. To determine if lithium was producing neuropil increases, quantitative three-dimensional magnetic resonance imaging studies were undertaken, which revealed that chronic lithium significantly increases total gray matter volume in the human brain of patients with manic– depressive illness. Together, these results suggest that a reconceptualization about the optimal long-term treatment of recurrent mood disorders is warranted. Optimal long-term treatment for these severe illnesses may only be achieved by the early use of agents with neurotrophic/neuroprotective effects, irrespective of the primary, symptomatic treatment. Biol
Psychiatry 2000;48:740 –754 © 2000 Society of Biological
Psychiatry
Key Words: Mood disorders, lithium, valproate, manic–
depressive illness, bcl-2, MAP kinase, neurite, neurotro-phic, N-acetylaspartate, gray matter
Introduction
M
anic– depressive illness (MDI) is a common, severe, chronic, often life-threatening illness (Goodwin and Jamison 1990). Suicide is the cause of death in 10 –20% of individuals with MDI, and the risks of suicide in MDI may be higher than those in unipolar depression. In addition to suicide, major mood disorders are also associated with many other deleterious health-related effects, and the costs associated with disability and premature death represent an economic burden of tens of billions of dollars annually in the United States alone (for a review, see Goodwin and Jamison 1990). Despite extensive research, the biochem-ical abnormalities underlying the predisposition to and the pathophysiology of MDI remain to be fully elucidated (Joffe and Young 1997). Although mood disorders have traditionally been regarded as “good prognosis diseases,” a growing body of data suggests that the long-term outcome for many patients is often much less favorable than previously thought. Indeed, according to the Global Burden of Disease Study, MDI is one of the leading causes of disability worldwide (Murray and Lopez 1997a, 1997b). In this context, although mood disorders were classically viewed as recurring conditions with essentially well periods between episodes, it has been increasingly recognized that interepisode recovery is incomplete in many patients, with a progressive decline in overall functioning (Goldberg and Harrow 1999).Are Mood Disorders Associated with
Structural Brain Changes?
In view of the deteriorating long-term clinical course observed in many patients (vide supra), it is not surprising that recent studies have been investigating potential struc-tural brain changes in mood disorders. It is noteworthy that although mood disorders have traditionally been concep-tualized as neurochemical disorders, there is now consid-erable literature support from a variety of sources demon-From the Laboratory of Molecular Pathophysiology, Department of Psychiatry &
Behavioral Neurosciences and Cellular and Clinical Neurobiology Program, Wayne State University School of Medicine, Detroit, Michigan.
Address reprint requests to Husseini K. Manji, M.D., Laboratory of Molecular Pathophysiology, National Institute of Mental Health, 10 Center Drive, 10/4N-222, MSC 1381, Bethesda MD 20892.
Received February 18, 2000; revised June 15, 2000; accepted June 23, 2000.
© 2000 Society of Biological Psychiatry 0006-3223/00/$20.00
strating that mood disorders are also associated with significant reductions in regional central nervous system (CNS) volume and cell numbers (both neurons and glia). One line of evidence comes from structural imaging studies, which have recently begun to provide important clues about the neuroanatomic basis of mood disorders. In toto, the majority of the volumetric neuroimaging studies have demonstrated an enlargement of third and lateral ventricles, as well as reduced basal ganglia and frontal cortical volumes (for reviews, see Drevets et al 1999; Sheline 2000). Within the frontal lobe, volumetric neuro-imaging studies have fairly consistently shown reduced volumes; in particular, recent volumetric magnetic reso-nance imaging (MRI) studies in familial bipolar depres-sives and familial unipolar depresdepres-sives have demonstrated reductions in the mean gray matter volume of approxi-mately 40% in the prefrontal cortex (PFC) ventral to the genu of the corpus callosum (Drevets 2000). Lending support to the structural neuroimaging literature are mul-tiple functional brain imaging studies that have shown abnormalities in metabolic rate and blood flow in the striatal, frontal, and temporal regions in mood disorders (for reviews, see Drevets et al 1999; Mayberg et al 2000). In addition to the accumulating neuroimaging evidence, several postmortem brain studies are now providing direct evidence for reductions in regional CNS volume and cell numbers (Table 1). Thus, studies have reported a layer-specific reduction in interneurons in the anterior cingulate cortex (Vincent et al 1997) and reductions in nonpyrami-dal neurons (;40% lower) in CA2 of the hippocampal formation in MDI subjects, as compared with control subjects (Benes et al 1998). Baumann and associates (Baumann and Bogerts, in press; Baumann et al 1999) reported reduced volumes of the left nucleus accumbens, the right putamen, and bilateral pallidum externum in postmortem brain samples obtained from patients with unipolar depression or MDI. Several recent postmortem studies of the PFC have also demonstrated reduced CNS volume and cell numbers in mood disorders. Rajkowska (1997) has used three-dimensional cell counting and mor-phological techniques to demonstrate decreased cortical and laminar thickness in MDI subjects completing suicide.
In thorough follow-up series from the same group, mor-phometric analysis of the density and size of cortical neurons in the dorsolateral PFC and orbitofrontal cortex revealed significant reductions in mood disorders. Addi-tionally, unexpected reductions in the glial cell number and density were also observed (Rajkowska 2000). Also in the PFC, a histologic study of area sg24 located in the subgenual PFC found striking reductions in glial cell numbers in patients with familial major depression (24% reductions) and MDI (41% reductions), as compared with control subjects (Drevets 2000). This striking finding is consistent with this group’s neuroimaging findings show-ing cortical volume loss in this same region on volumetric MRI in a similar diagnostic group. Most recently, Drevets and associates have investigated the effects of medications on the volumetric changes observed in the subgenual PFC; in view of lithium and valproate’s robust effects on the levels of the cytoprotective protein bcl-2 in the frontal cortex (discussed in detail later), Drevets and associates reanalyzed their data to determine if there was an effect of medication use. Entirely consistent with a neurotrophic/ neuroprotective role for bcl-2, they found that the patients treated with lithium or valproate exhibited subgenual PFC volumes that were significantly higher than the volumes in non–lithium- or valproate-treated patients and not signif-icantly different from those of control subjects.
Together, the majority of the data from the neuroimag-ing studies and the growneuroimag-ing body of postmortem evidence present a convincing case that there is indeed a reduction in regional CNS volume, accompanied by a reduction in cell numbers in at least a subset of patients with mood disorders. It remains to be elucidated if these findings represent neurodevelopmental abnormalities, disease pro-gression that fundamentally involves loss/atrophy of glia
and neurons, or the sequelae of the biochemical changes
(e.g., in glucocorticoid levels) accompanying repeated affective episodes per se. In support of the latter, chronic stress or glucocorticoid administration has been demon-strated to produce atrophy or even death of vulnerable hippocampal neurons in rodents and primates, and MRI studies have also revealed reduced hippocampal volumes in patients with Cushing disease and posttraumatic stress Table 1. Postmortem Morphometric Studies Suggesting Cell Death and/or Atrophy in Mood Disorders
●™parahippocampal cortex size in suicide (Altshuler et al 1990)
●Layer-specific™in interneurons in the anterior cingulate cortex in BD (Vincent et al 1997)
● ™nonpyramidal neurons in the CA2 region in BD (and schizophrenia; Benes et al 1998) ●™cortical/laminar thickness in BD (suicide; Rajkowska 1997)
●™volumes of (L) nucleus accumbens, bilateral basal ganglia (Baumann and Bogerts, in press; Baumann et al 1999) ●™density and size of cortical neurons in the DLPFC and orbitofrontal cortices in mood disorders (Rajkowska 2000) ●™glial cell number and density in the DLPFC in mood disorders (Rajkowska 2000)
●™glia (but not neurons) in the subgenual PFC in familial MDD (24% reductions) and BD (41% reductions; Drevets 2000)
disorder. The distinction between repeated episodes and disease progression is not a trivial one, since it may be possible to utilize therapeutic strategies to reduce the cell loss/atrophy associated with disease progression, even if these agents do not have a major impact on the intensity/ frequency of the affective episodes per se.
Can Disease-Related CNS Cell Death or
Atrophy Be Attenuated or Reversed?
As discussed above, it is now clear that mood disorders are often associated with cell loss and atrophy; these structural brain changes may contribute to the poor long-term outcome observed in many patients. Thus, the develop-ment of strategies to attenuate or even reverse regional brain cell loss/atrophy may be critical for improved long-term outcome. Are such therapeutic strategies even plausible? The dependence of neuronal survival on spe-cific “survival factors” and genetic programs represents an intricate and elegant scheme by which much of the establishment, molding, and refining of neuronal circuitry occurs physiologically. A growing body of data has shown that many of the same pathways may also be involved in the cell death and atrophy that occurs pathologically in certain disorders. With the realization that disease-related cell death may arise from aberrantly activated gene-directed processes and/or the absence of critical trophic signals, the loss or atrophy of large numbers of cells in the CNS no longer has to be accepted as an unavoidable fate. In addition to the identification of specific cellular path-ways that may be involved in regulating cell survival, it has recently been demonstrated that neurogenesis occurs in the adult human brain (Eriksson et al 1998; Kemper-mann and Gage 1999). The localization of pluripotent progenitor cells and neurogenesis occurs in restricted brain regions. The greatest density of new cell birth is observed in the subventricular zone and the subgranular layer of the hippocampus. Cells born in the subventricular zone mi-grate largely to the olfactory bulb, and those in the subgranular zone into the granule cell layer. A recent study has demonstrated that new neurons, originating from the subventricular zone, are found also in areas of the associ-ation cortex of nonhuman primates (Gould et al 1999b). The newly generated neurons send out axons and appear to make connections with surrounding neurons, indicating that they are capable of integrating into the appropriate neuronal circuitry in the hippocampus and cerebral cortex. Neurogenesis in the hippocampus is increased by enriched environment, exercise, and hippocampal-dependent learn-ing (Gould et al 1999a, 2000; Kempermann et al 1997; van Praag et al 1999). Upregulation of neurogenesis in re-sponse to these behavioral stimuli and the localization of this process to the hippocampus and association cortex
have led to the proposal that new cell birth is involved in learning and memory (Gould et al 1999a, 2000).
Recent studies have shown that decreased neurogenesis occurs in response to both acute and chronic stress (Gould et al 2000). Removal of adrenal steroids (i.e., adrenalec-tomy) increases neurogenesis, and treatment with high levels of glucocorticoids reproduces the downregulation of neurogenesis that occurs in response to stress. Aging also influences the rate of neurogenesis. Although neurogen-esis continues into late life, the rate is significantly reduced. The decreased rate of cell birth could result from upregulation of the hippocampal–pituitary–adrenal axis and higher levels of adrenal steroids that occur in later life. Lowering glucocorticoid levels in aged animals restores neurogenesis to levels observed in younger animals, indi-cating that the population of progenitor cells remains stable but is inhibited by glucocorticoids (Cameron and McKay 1999). These observations raise the interesting possibility that corticotropin-releasing factor antagonists, currently being developed for the treatment of mood and anxiety disorders, may have particular utility in the treat-ment of elderly depressed patients. Also of potential relevance to our understanding of the neurobiology and treatment of mood disorders, ovariectomy decreases the proliferation of new cells in the hippocampus, effects that are reversed by estrogen replacement (Gould et al 2000). The rate of neurogenesis fluctuates over the course of the estrus cycle in rodents, and the total rate of cell birth is higher in female rodents. In addition to potentially playing a role in the beneficial cognitive effects of estrogen, the regulation of neurogenesis by this gonadal steroid may also provide important clues about certain sexually dimor-phic characteristics of mood disorders. These recent ad-vances in our understanding of the factors that regulate cell survival and neurogenesis have led to considerable excitement about the prospect of pharmacologic interven-tion in the adult mammalian brain (Chen et al 2000; Kempermann and Gage 1999).
The Discovery of bcl-2 as a Therapeutically
Relevant Target for the Actions of Lithium
and Valproate: The Successful Application
of a Concerted mRNA Differential Display
Strategy to Identify Novel Target Genes
com-plex alterations in basal and stimulated DNA binding to activator protein 1 (AP-1) transcription factors (Asghari et al 1999; Chen et al 1999a; Jope 1999; Manji et al 2000a; Ozaki and Chuang 1997). Together, these data suggest that lithium and valproate, via their effects on the AP-1 family of transcription factors, may bring about strategic changes in gene expression in critical neuronal circuits, effects that may ultimately underlie its efficacy in the treatment of a very complex neuropsychiatric disorder. Although many genes that are the targets of long-term lithium and val-proate have indeed been identified, it has been estimated that;10,000 –15,000 genes may be expressed in a given cell at any time, and thus additional, novel methodologies are clearly required to study the complex pattern of gene expression changes induced by chronic drug treatment. One methodology that has been successfully utilized to identify the differential expression of multiple genes (e.g., in pathologic vs. normal tissue, or in control vs. treated tissue) is reverse transcription polymerase chain reaction messenger RNA (mRNA) differential display (Liang et al 1995). Using this method, Wang and Young (1996) were the first to make the novel observation that lithium increased 29,39-cyclic nucleotide 39-phosphodiesterase mRNA levels in C6 glioma cells. Subsequent studies have also utilized this methodology to identify novel genes that may be the targets for the actions of mood stabilizers. A major problem inherent in neuropharmacologic research, however, is the dearth of phenotypic changes clearly associated with treatment response, particularly for mood-stabilizing agents (Ikonomov and Manji 1999). In the absence of clear-cut phenotypic changes, we have at-tempted to overcome this experimental hurdle by utilizing paradigms that involve the identification of common long-term molecular targets of structurally dissimilar mood-stabilizing agents (lithium and valproate) when administered chronically in vivo (Chen et al 1999c). These are two distinct agents that undoubtedly also exert a variety of dissimilar effects; however, the identification of genes affected by both agents, when administered in a therapeutically relevant paradigm, suggests that such mo-lecular changes may play a role in the common therapeutic effects of these agents. One of the genes whose expression was markedly increased by the treatments is the transcrip-tion factor PEBP2b(GenBank Accession No. AF087437; discussed in Chen et al 1999c). It was further demon-strated that PEBP2b’s function (DNA binding of the PEBP2 ab complex) was also clearly increased in the frontal cortex by both lithium and valproate, but not by chronic amphetamine or benzodiazepine. One critical gene whose expression is known to be regulated by PEBP2bis the major neuroprotective protein bcl-2; it was subse-quently demonstrated that chronic treatment of rats with both agents resulted in a doubling of bcl-2 levels in the
frontal cortex. Furthermore, immunohistochemical studies showed that chronic treatment of rats with lithium or valproate resulted in a marked increase in the number of bcl-2–immunoreactive cells in layers II and III of the frontal cortex. Interestingly, the importance of neurons in layers II–IV of the frontal cortex in mood disorders has recently been emphasized, since primate studies have indicated that these are important sites for connections with other cortical regions, and major targets for subcor-tical input (Rajkowska et al 1999). Chronic lithium also markedly increased the number of bcl-2–immunoreactive cells in the dentate gyrus and striatum (Manji et al 1999); detailed immunohistochemical studies following chronic valproate treatment are currently underway. It was subse-quently demonstrated that lithium also increases bcl-2 levels in C57BL/6 mice (Chen et al 2000), and in human neuroblastoma SH-SY5Y cells in vitro (Manji et al 2000b); our demonstration of a lithium-induced increase in bcl-2 levels has also been convincingly replicated in rat cerebellar granule cells (Chen and Chuang 1999) in recent studies. This latter study was undertaken to investigate the molecular and cellular mechanisms underlying the neuro-protective actions of lithium against glutamate excitotox-icity (vide infra). These investigators found that lithium produced a remarkable increase in bcl-2 protein and mRNA levels. Moreover, lithium has very recently been demonstrated to reduce the levels of the pro-apoptotic protein p53 in both cerebellar granule cells (Chen and Chuang 1999) and SH-SY5Y cells (Lu et al 1999). Thus, overall the data clearly show that chronic lithium robustly increases the levels of the neuroprotective protein bcl-2 in areas of the rodent frontal cortex, hippocampus, and striatum in vivo, and in cultured cells of both rodent and
human neuronal origin in vitro; furthermore, at least in
cultured cell systems, lithium has also been demonstrated to reduce the levels of the proapoptotic protein p53.
in the process of apoptosis, and studies have shown that mitochondria undergo major changes in membrane integ-rity before classic signs of apoptosis become manifest, leading to a disruption of the inner transmembrane poten-tial (DCm) and the release of intermembrane proteins
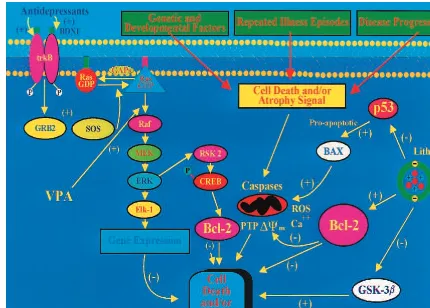
through the outer membrane (Figure 1). Chemically in-duced opening or closing of the permeability transition pore (also called mitochondrial megachannel) can induce or prevent apoptosis, respectively. bcl-2 acts on mitochon-dria to stabilize membrane integrity and to prevent open-ing of the permeability transition pore, and has been
shown to protect neurons from a variety of insults both in vitro and in vivo (for reviews, see Adams and Cory 1998; Bruckheimer et al 1998; Table 2). Overexpression of bcl-2 in transgenic mice has been shown to prevent motor neuron and retinal ganglion death, to protect cells from the deleterious effects of N-methyl-4-phenyl-1,2,3,6-tetrahy-dropyridine (MPTP) or focal ischemia, to protect photo-receptor cells from inherited retinal degeneration, and to prolong survival and attenuate motor neuron degeneration in a transgenic animal model of amyotrophic lateral sclerosis (discussed in Adams and Cory 1998; Bruck-Figure 1. Neurotrophic and neuroprotective effects of lithium and valproate (VPA). This figure depicts the mechanisms by which lithium and VPA exert neurotrophic and neuroprotective effects in the long-term treatment of mood disorders. Genetic/neurodevel-opmental factors, repeated affective episodes (and likely elevations of glucocorticoids), and illness progression may all contribute to the volumetric reductions and cell death/atrophy observed in mood disorders. bcl-2 attenuates apoptosis by sequestering proforms of death-driving cysteine proteases (called caspases); by preventing the release of mitochondrial apoptogenic factors such as calcium, cytochrome c, and apoptosis-inducing factor into the cytoplasm; and by enhancing mitochondrial calcium uptake. Mitochondria undergo major changes in membrane integrity before classic signs of apoptosis become manifest, leading to a disruption of the inner transmembrane potential (DCm) and the release of intermembrane proteins through the outer membrane; bcl-2 acts on mitochondria
heimer et al 1998; Manji et al 1999; Merry and Korsmeyer 1997 and references therein).
It is also noteworthy that recent studies have demon-strated that bcl-2 may mediate many of the downstream effects of several neurotrophic factors. Neurotrophic fac-tors (e.g., nerve growth factor and brain-derived neurotro-phic factor) are now known to promote cell survival by binding to membrane receptors (such as Trk A and Trk B) and regulating intracellular signal transduction pathways that can control apoptosis. The signal transduction cas-cades that have been identified include the mitogen-activated protein (MAP) kinase cascade and the phos-photidylinositol-3 kinase (PI-3K)/Akt pathway (Segal and Greenberg 1996; Tao et al 1998). Recent studies have demonstrated that the activation of the MAP kinase pathway can inhibit apoptosis by inducing the phosphor-ylation of Bad and increasing the expression of bcl-2, the latter effect likely involving the cAMP response element–
binding protein (CREB; Bonni et al 1999; Riccio et al 1999). Phosphorylation of Bad occurs via activation of a downstream target of the MAP kinase cascade, ribosomal S-6 kinase (Rsk). Rsk phosphorylates Bad and thereby promotes its inactivation. Activation of Rsk also mediates the actions of the MAP kinase cascade and neurotrophic factors on the expression of bcl-2. Rsk can phosphorylate CREB, and this leads to induction of bcl-2 gene expres-sion. Moreover, in addition to its neuroprotective effects, bcl-2 overexpression has also been shown to promote
regeneration of axons in the mammalian CNS and
regu-late to neurite sprouting (Chen et al 1997). Thus, it has been convincingly argued that pharmacologic means of increasing CNS bcl-2 levels may represent a very effective therapeutic strategy for the treatment of many neurode-generative diseases (Chen et al 1997).
Does Inhibition of Glycogen Synthase
Kinase 3
b
(GSK-3
b
) Play a Role in the
Therapeutic Effects of Mood Stabilizers?
Klein and Melton (1996) were the first to make the seminal observation that lithium, at therapeutically rele-vant concentrations, is an inhibitor of GSK-3b. Glycogen synthase kinase 3b not only controls developmental pat-terns in diverse organisms (including mammals), but also plays an important role in the mature CNS, by regulating various cytoskeletal processes as well as long-term nuclear events via phosphorylation of c-jun, nuclear translocation of b-catenin, and nuclear export of nuclear factor of activated T cells (for reviews, see Dale 1998; Jope 1999; Willert and Nusse 1998). Glycogen synthase kinase 3bhas also been implicated in regulating the phosphorylation of
tau andb-catenin, both of which have been implicated in certain types of disease-related neuronal death (discussed in Dale 1998; Manji et al 1999; Nishimura et al 1999; Willert and Nusse 1998; Zhang et al 1998). Overexpres-sion of a catalytically active GSK-3 has been shown to induce apoptosis of PC12 cells, and dominant-negative GSK-3 prevents apoptosis following inhibition of PI-3K (Pap and Cooper 1998). Furthermore, overexpression of GSK-3b(to levels 3.5 times that in control cells) has been demonstrated to potentiate staurosporine-induced caspase activation as well as morphological changes indicative of apoptosis (Bijur et al 2000). Thus, inhibition of GSK-3b
by lithium may also afford protection against the cell death induced by certain stimuli (Hetman et al 2000). In this context, several recent studies have found that inhibition of GSK-3b by lithium reduces tau phosphorylation, ef-fects that likely occur to some degree at therapeutically relevant lithium concentrations (see Jope 1999 for an excellent discussion). Although many of the studies have utilized lithium concentrations in excess of those utilized Table 2. Neuroprotective Effects of bcl-2 and Lithium
bcl-2
●Protects against the lethal effects of a variety of reactive oxygen species generating insults
●Protects against MPTP and AMPA neurotoxicity ●Protects against growth factor deprivation ●Protects against the effects of ionizing radiation ●Protects against glucocorticoid toxicity ●Reduces neuropathology after focal ischemia ●Reduces neuropathology after traumatic brain injury ●Prevents axotomy-induced motor neuron death
●Attenuates motor neuron degeneration in a transgenic animal model of ALS
●Promotes regeneration of axons in the mammalian central nervous system
Lithium
●Protects cultured neurons against glutamate and NMDA-induced cell death
●Protects cerebellar granule cells from KCl deprivation and age- or drug-induced apoptosis
●Protects PC12 cells after nerve growth factor deprivation ●Protects SH-SY5Y cells from ouabain toxicity
●Delays radiation-induced apoptosis in mice
●Protects SH-SY5Y cells from Ca11and MPP1toxicity ●Protects cells from the deleterious effects of glycogen synthase
kinase 3boverexpression coupled to staurosporine addition ● Protects cultured rat cortical neurons fromb-amyloid toxicity ● Reduces behavioral deficits and choline acetyltransferase activity
induced by cholinergic lesions
● Reduces MCA occlusion-induced infarct size and neurologic deficits ● Protects neurons in the striatum from quinolinic acid toxicity
ALS, amyotrophic lateral sclerosis; NMDA, N-methyl-D-aspartate; MPP1,
therapeutically, the available data suggest that lithium, at concentrations of ;1 mmol/L does, indeed, reduce tau phosphorylation (Hong et al 1997; Jope 1999; Lovestone et al 1999; Munoz-Montano et al 1997). In view of the important role of GSK-3b in regulating cyctoskeletal events and cell survival, a study was undertaken to determine if valproate also regulates GSK-3b; it was found that valproate also significantly inhibited GSK-3b
at therapeutically relevant concentrations, and consistent with GSK-3b inhibition, incubation of SH-SY5Y cells with valproate resulted in a significant time-dependent increase in both cytosolic and nuclear b-catenin levels (Chen et al 1999b). Another independent laboratory has recently also demonstrated that valproate increasesb -cate-nin levels and increases the expression of a reporter gene driven by b-catenin/lymphoid enhancer factor transcrip-tion factor (P.S. Klein, personal communicatranscrip-tion, March 2000). Most recently, it has been demonstrated that the chronic (3– 4 week) administration of lithium and val-proate also increase b-catenin levels in the rodent brain (G. Chen and H.K. Manji, unpublished observations), compatible with inhibition of GSK-3bduring chronic in vivo administration of the agents under therapeutic paradigms.
Neuroprotective Effects of Lithium:
Compelling Preclinical Evidence
Lithium’s robust effects on bcl-2 and GSK-3b in the mature CNS suggest that it may possess significant
neu-roprotective properties. Indeed, several studies conducted before the identification of bcl-2 or GSK-3bas targets for lithium’s actions had already demonstrated
neuroprotec-tive properties of lithium (Alvarez et al 1999; D’Mello et
al 1994; Grignon et al 1996; Inouye et al 1995; Li et al 1994; Pascual and Gonzalez 1995; Volonte and Ruken-stein 1993). The protective effects of lithium have been investigated in a number of in vitro studies, in particular using rat cerebellar granule cells, PC12 cells, and human neuroblastoma SH-SY5Y cells. In these studies, lithium has been shown to protect against the deleterious effects of glutamate, N-methyl-D-aspartate receptor activation, low
potassium, and toxic concentrations of anticonvulsants (Table 2). Lithium also protects PC12 cells from serum/ nerve growth factor deprivation (Volonte and Rukenstein 1993), protects both PC12 cells and human neuroblastoma SH-SY5Y cells from ouabain toxicity (Li et al 1994), and protects SH-SY5Y cells from both thapsigargin (which mobilizes intracellular Ca11) and 1-methyl-4-phenylpyri-dinium ion (MPP1)–induced cell death (Figure 2). Most recently, lithium has been shown to protect cultured neurons frombamyloid–induced cell death (Alvarez et al 1999) and to protect against the deleterious effects of
GSK-3boverexpression coupled to staurosporine addition (Bijur et al 2000; Table 2).
In addition to the demonstration of protective effects in vitro, a number of studies have also investigated lithium’s neuroprotective effects in vivo. In this con-text, studies have investigated the effects of lithium on the biochemical and behavioral manifestations of exci-totoxic lesions of the cholinergic system (Arendt et al 1999; Pascual and Gonzalez 1995). These studies have demonstrated that lithium pretreatment attenuates both the behavioral deficits (passive avoidance and ambula-tory behavior) and the reduction in choline acetyltrans-ferase (ChAT) activity by forebrain cholinergic system lesions (Pascual and Gonzalez 1995). In a study that may have implications for the treatment of Alzheimer’s disease, rats received ibotenic acid lesions of cholin-ergic basal forebrain nuclei, resulting in a 30 – 40% depletion of both ChAT and acetylcholinesterase (AChE) activity (Arendt et al 1999). Lithium as well as tetrahydroaminoacridine, given separately either before or after the development of the lesion, had small but significant effects on the recovery of cortical ChAT and AChE activity. Intriguingly, when applied in combina-tion the drugs clearly showed synergistic effects; how-ever, considerable caution is required in the extrapola-tion of these results to the treatment of humans, since the coadministration of lithium and cholinesterase in-hibitors has been shown to be capable of inducing seizures. In another study investigating lithium’s effects against excitoxic insults, it was demonstrated that lithium attenuated the kainic acid–induced reduction in glutamate decarboxylase levels and [3H]
D-aspartate
Furthermore, Datta et al (1997) have demonstrated that Akt phosphorylation of BAD (a proapoptotic member of the bcl-2 family) blocks BAD-induced death of primary neurons. These results suggest that lithium’s effects on Akt-1 may also contribute to neuroprotective effects; however, such a contention awaits the demonstration of lithium-induced activation of Akt-1 in the CNS in vivo.
Does Lithium Affect Neurogenesis?
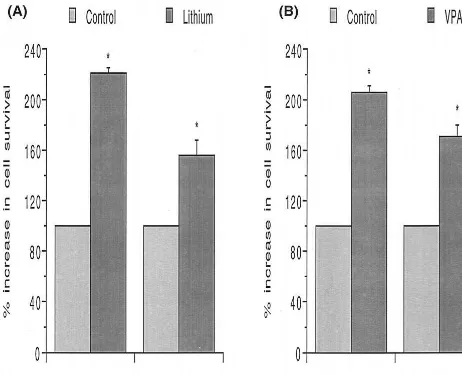
As discussed already, it has been shown, with a method for labeling cell division directly in the adult human brain, that the dentate gyrus (an area where robust lithium-induced increases in bcl-2 levels are observed) can produce new neurons during adulthood in humans. A large number of the newborn daughter cells are known to die rapidly, likely via apoptosis (Kempermann and Gage 1999). Thus, increasing bcl-2 levels could enhance the survival of the newborn cells, Figure 2. Neuroprotective effects of lithium and valproate (VPA) in vitro. Human SH-SY5Y neuroblastoma cells were incubated with lithium (1.0 mmol/L) or VPA (1.0 mmol/L) for 3 days and were then exposed to two different toxins—thapsigargin (which mobilizes intracellular calcium; 0.5 mmol/L for 16 hours) or MPP1(25mmol/L for 16 hours) The mitochondrial dehydrogenase activity that
allowing them to differentiate into neurons. Additionally, bcl-2 has been shown to have robust effects on the
regener-ation of CNS axons (Chen et al 1997). In view of bcl-2’s
major neuroprotective and neurotrophic role, a study was undertaken to determine if lithium, administered at therapeu-tically relevant concentrations, affects neurogenesis in the adult rodent brain. Kempermann and Gage (1999) have suggested the use of neurogenesis to refer to a series of events (including proliferation of a neuronal precursor or stem cell, and survival of the daughter cells) that result in the appearance of a new neuron. To investigate the effects of chronic lithium on neurogenesis, mice were treated with “therapeutic” lithium (plasma levels 0.976 0.20 mmol/L), for;4 weeks. After treatment with lithium for 14 days, the mice were administered single doses of bromodeoxyuridine (BrdU), a thymidine analog that is incorporated into the DNA of dividing cells, for 12 consecutive days. Lithium treatment continued throughout the duration of the BrdU administra-tion. Following BrdU immunohistochemistry (Chen et al 2000), three-dimensional cell counting was performed using a computer-assisted image analysis system (Rajkowska 2000). This system is based on the optical dissector method and estimates the number of cells independent of section thickness and cell shape. We found that chronic lithium administration does, indeed, result in an increase in the number of BrdU-positive cells in the dentate gyrus (Chen et al 2000). Moreover, approximately two thirds of the BrdU-positive cells also double stained with the neuronal marker NeuN, confirming their neuronal identity. Double staining of BrdU and bcl-2 was also observed, and studies using bcl-2 transgenic animals are currently underway to delineate the role of bcl-2 overexpression in the enhanced hippocampal neurogenesis observed.
Neurotrophic and Neuroprotective Effects of
Valproate
Valproate’s effects on bcl-2 and GSK-3bsuggest that this mood stabilizer may also possess neuroprotective/neuro-trophic properties. Additionally, it has been recently dem-onstrated that valproate increases the expression of the molecular chaperone GRP78, a protein that binds Ca11in the endoplasmic reticulum, and protects cells from the deleterious effects of damaged proteins (Wang et al 1999). Although it is not as extensively studied as lithium, a growing body of data suggests that valproate does, indeed, exert neuroprotective effects (Bruno et al 1995; Mark et al 1995; Mora et al 1999). We have found that, similar to what has been observed in vivo, incubation of human neuroblastoma SH-SY5Y cells with either lithium (1.0 mmol/L) or valproate (0.6 –1.0 mmol/L) significantly increases the levels of bcl-2. These results suggest that the in vivo increases in bcl-2 levels may be due, at least in
part, to direct cellular effects of these agents (rather than secondarily due to alterations in synaptic input or long-loop feedback systems that are present in the in vivo condition). We have therefore utilized the SH-SY5Y model system to investigate valproate and lithium’s pro-tective effects. SH-SY5Y cells were incubated with lith-ium (1.0 mmol/L) or valproate (0.6 mmol/L) for 3 days. Cells were then exposed to two different toxins—thapsi-gargin (which mobilizes intracellular calcium; 0.5 mmol/L for 16 hours) or MPP1 (25 mmol/L for 16 hours). The mitochondrial dehydrogenase activity that cleaves 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide was used to determine cell survival in a quantitative colorimetric assay. It was found that lithium and valproate treatment exerted significant protective effects against both toxins (Figure 2).
Robust Effects of Valproate on MAP
Kinases
Although a growing body of data has shown that valproate increases AP-1 DNA binding activity and AP-1–mediated gene expression (vide supra), the mechanisms underlying these effects have not been fully elucidated. In this context, MAP kinases play a key role in the regulation of the AP-1 family of transcription factors (Gutkind 1998). Mitogen-activated protein kinases transmit extracellular signals to the nucleus, where the transcription of specific genes is induced by the synthesis, phosphorylation, and activation of transcription factors. Three distinct MAP kinase signal transduction pathways have been identified in mammalian cells, leading to activation of the MAP kinases— extracellular signal–regulated kinases (ERKs), c-Jun NH2-terminal kinases, and p38 (Gutkind 1998). Mitogen-activated protein kinases are abundantly present in the brain, and in recent years a broad role for the MAP kinase cascade in regulating gene expression in long-term forms of synaptic plasticity has been demonstrated (En-glish and Sweatt 1997; Martin et al 1997; Roberson et al 1999). Thus, MAP kinases play important physiologic roles in the mature CNS and have been postulated to represent important targets for the actions of CNS-active agents (Nestler 1998; Yuan et al 1999; G. Chen et al, unpublished data).
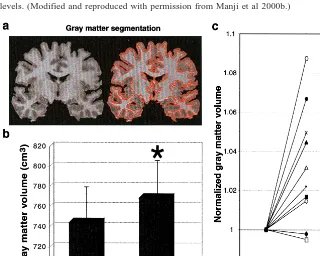
Figure 4. Brain gray matter volume is increased following 4 weeks of lithium administration at therapeutic levels in manic– depressive illness (MDI) patients.
(a) A slice of the three-dimensional
volu-metric magnetic resonance imaging (MRI) data that was segmented by tissue type using quantitative methodology to deter-mine tissue volumes at each scan time point. Brain tissue volumes were exam-ined using high resolution three-dimen-sional MRI (124 1.5-mm–thick coronal T1-weighted spoiled gradient echo
nerve growth factor and brain-derived neurotrophic factor [BDNF]) and to promote neurite outgrowth (Finkbeiner 2000; Segal and Greenberg 1996), we investigated val-proate’s effects on the morphology of SH-SY5Y cells in detail. SH-SY5Y cells exposed to valproate’s (1.0 mmol/L) in serum-free media for 5 days exhibited prom-inent growth cones and long neurites. Growth cone– associated protein 43 (GAP-43) is a protein expressed at elevated levels during neurite growth during development or regeneration, and a greater than threefold increase in GAP-43 levels was observed after 5-day valproate expo-sure (G. Chen et al, unpublished data). bcl-2 is another protein known to play a key role in neuronal development and neurite growth, and 5-day valproate exposure resulted in greater than fivefold increase in bcl-2 levels. To further analyze the valproate-induced morphological changes, SH-SY5Y cells were seeded at a very low density and treated with valproate in serum-free media for 1 to 9 days. Valproate (0.5 mmol/L) dramatically increased neurite length approximately ninefold after 4-day incubation, and
;14-fold after 9-day incubation. In view of valproate’s apparent trophic effects, SH-SY5Y cells were grown in the presence of a therapeutic concentration of valproate
with-out any additional neurotrophic factors. Remarkably, cells
grown in the presence of valproate (but without other neurotrophic factors) continued to grow well for longer than 40 days.
As discussed, a variety of neurotrophins activate the ERK pathway via cell surface tyrosine kinase receptors (e.g., trkB), and ERK pathways are known to play a major role in neurotrophin-induced cell differentiation and neu-rite growth (Finkbeiner 2000; Segal and Greenberg 1996). Interestingly, recent studies have demonstrated that the chronic administration of a variety of antidepressants increases the expression of BDNF, neurotrophin-3, and trkB (Duman et al 1997; Smith et al 1995). It is thus noteworthy that valproate activates ERKs and promotes neurite growth in SH-SY5Y cells, effects that are charac-teristic of neurotrophic factors; these results, as well as the dramatic prolongation of the survival of SH-SY5Y cells grown in the absence of other trophic factors, suggests that valproate possesses neurotrophic properties and may thus have utility in the treatment of other neuropsychiatric disorders.
Can Lithium’s
Neurotrophic/Neuroprotective Effects Be
Demonstrated Longitudinally in Humans in
the CNS In Vivo?
A clinical study was recently undertaken to determine if lithium also exerts neurotrophic/neuroprotective effects in the human brain in vivo. Proton magnetic resonance spectroscopy (MRS) is a tool that provides a noninvasive window to functional brain neurochemistry.
N-Acetyl-aspartate (NAA) is one of the many neurochemical com-pounds that can be quantitatively assessed via MRS.
N-Acetyl-aspartate is the predominant resonance in the
proton MRS spectrum of the normal adult human brain, and although the functional role of this amino acid has not been definitively determined, NAA is a putative neuronal marker (Birken and Oldendorf 1989), localized to mature neurons and not found in mature glial cells, cerebrospinal fluid, or blood. A relative decrease in this compound may reflect decreased neuronal viability, neuronal function, or neuronal loss (for an excellent recent review of NAA, see Tsai and Coyle 1995). In a prospective longitudinal study, we have utilized quantitative in vivo proton MRS to test the hypotheses that 1) similar to the preclinical findings in the rodent brain and in human neuronal cells in culture, chronic lithium increases neuronal viability/function in the
human brain in vivo, as evidenced by increased CNS
levels of NAA, and 2) putative lithium-induced increases in NAA levels are positively correlated with regional brain gray matter content.
Proton MR spectra were acquired from 8-cm3regions of interest (ROIs) in the frontal, temporal, parietal, and occipital lobes, with an acquisition time of 5 min per ROI (stimulated echo acquisition mode [STEAM] pulse se-quence; echo time5 30 msec, modulation time 5 13.7 msec, repetition time5 2000 msec; Moore et al 1999). Two trained individuals analyzed the in vivo nuclear MR data with MRUI-VARPRO time domain spectral analysis software (deBeer et al 1992; van den Boogaart et al 1994); these individuals were blind to the study information and to each other’s results. After extensive validation of this method for in vivo measurement of regional NAA con-centration, we have begun to apply this methodology in our studies of MDI patients and healthy volunteers under-going lithium administration. It was found that chronic lithium administration at therapeutic doses increases NAA concentration in the human brain in vivo (Moore et al 2000). These findings provide intriguing indirect support for the contention that, similar to the findings observed in the rodent brain and in human neuronal cells in culture, chronic lithium increases neuronal viability/function in the human brain. Furthermore, a striking ;.97 correlation between lithium-induced NAA increases and regional voxel gray matter content was observed (Moore et al 2000), thereby providing evidence for colocalization with the regional specific bcl-2 increases observed (e.g., gray vs. white matter) in the rodent brain cortices. These results suggest that chronic lithium may not only exert robust neuroprotective effects (as has been demonstrated in a variety of preclinical paradigms), but also exerts
neuro-trophic effects in humans (Figure 3).
and thus to increased brain gray matter volume in MDI patients. In this clinical research investigation we exam-ined brain tissue volumes using high resolution three-dimensional MRI (124 1.5-mm–thick coronal T1-weighted
SPGR images) and validated quantitative brain tissue segmentation methodology to identify and quantify the various components by volume, including total brain white and gray matter content. Measurements were made at baseline (medication free, after a minimum 14-day wash-out) and then repeated after 4 weeks of lithium at thera-peutic doses. This study revealed an extraordinary finding that chronic lithium significantly increases total gray
matter content in the human brain of patients with MDI
(Moore et al, in press; Figure 4). No significant changes were observed in brain white matter volume or in quanti-tative measures of regional cerebral water content, thereby providing strong evidence that the observed increases in gray matter content are likely due to neurotrophic effects as opposed to any possible cell swelling and/or osmotic effects associated with lithium treatment. A finer grained subregional analysis of this brain imaging data is ongoing. To our knowledge no other studies have examined the effects of chronic lithium treatment on brain tissue volume in MDI patients; however, the cross-sectional study by Drevets and associates demonstrating that lithium- or valproate-treated patients exhibited subgenual PFC gray matter volumes that were significantly higher than those of non–lithium- or non–VPA-treated patients, and the dem-onstration of gray matter–specific NAA increases with chronic lithium adds indirect support to these intriguing findings. Since it is believed that the majority of neuron-specific NAA is localized to the neurites rather than the cell body (Birken and Oldendorf 1989), the observed increase in NAA is likely due to expansion of neuropil content (Figure 3). Taken together, these exciting new results support the contention that lithium does indeed exert neurotrophic/neuroprotective events in the human brain in vivo.
Concluding Remarks
The evidence demonstrating the neurotrophic effects of lithium and valproate, the enhancement of hippocampal neurogenesis in the adult mammalian brain, and the growing appreciation that mood disorders are associated with cell loss and atrophy suggest that these effects may be very relevant for the long-term treatment of mood disor-ders. It should be emphasized that it is unclear if these neurotrophic effects are related to the acute treatment of mania or depression; it is quite likely that this enhance-ment of “cellular resiliency” is more related to long-term prophylactic effects and illness-related global deteriora-tion. Does the long-term administration of these agents
actually retard disease- or affective episode–induced cell loss or atrophy? As articulated, the distinction between
disease progression and affective episodes per se is an important one, since it is quite possible that the neurotro-phic effects of lithium or valproate may even be indepen-dent of their ability to treat or prevent affective episodes. There are currently no longitudinal studies that we are aware of that can fully address this question, but this is clearly a very important and fundamental issue worthy of investigation. The findings that lithium administration increases brain NAA levels and gray matter volumes, as well as the cross-sectional study demonstrating “normal-ized” subgenual PFC volumes in lithium- and valproate-treated patients, do provide indirect support for such a contention. Longitudinal studies investigating the poten-tial long-term neurotrophic effects (using serial volumetric MRI scans or MRS quantitation of regional NAA levels, for example) of mood stabilizers are clearly warranted. The evidence also suggests that early and potentially sustained treatment may be necessary to adequately pre-vent many of the deleterious long-term sequellae associ-ated with mood disorders. Although data suggest that hippocampal atrophy in depression is related to illness duration (Sheline 2000), it is currently not clear if the volumetric and cellular changes that have been observed in other brain areas (most notably the frontal cortex) are related to affective episodes per se. Indeed, some studies have observed reduced gray matter volumes and enlarged ventricles in mood disorder patients at first onset (Hira-yasu et al 1998; Strakowski et al 1993). At this point, it is unclear if the cell death and atrophy in mood disorders occur because of the magnitude and duration of the biochemical pertubations, an enhanced vulnerability to the deleterious effects of these pertubations (due to genetic factors and/or early life events), or a combination thereof (Fig 1). In conclusion, emerging results from a variety of clinical and preclinical experimental and naturalistic par-adigms suggest that a reconceptualization about the patho-physiology, course, and optimal long-term treatment of recurrent mood disorders is warranted. Optimal long-term treatment for these severe illnesses may only be achieved by the early use of agents with neurotrophic/neuroprotec-tive effects, irrespecneurotrophic/neuroprotec-tive of the primary, symptomatic treatment. Such treatment modalities, via their effects on molecules involved in cell survival and cell death path-ways, such as CREB, BDNF, bcl-2, and MAP kinases (Figure 1) would be envisioned as enhancing neuroplas-ticity and cellular resilience and modulating the long-term course of these devastating illnesses.
Schizophrenia and Depression, and Joseph Young Sr. Research grants. Outstanding editorial assistance was provided by Ms. Celia Knobelsdorf. Space limitations necessitate our citing of review articles in many cases. A full reference list upon which this article was based is available upon request.
Aspects of this work were presented at the conference “Depression in the Twenty-First Century: New Insights into Drug Development and Neurobiology,” February 21–22, 2000, Dana Point, California. The conference was sponsored by the Society of Biological Psychiatry through an unrestricted educational grant provided jointly by Pharmacia & Upjohn and Janssen Pharmaceutica.
References
Adams JM, Cory S (1998): The Bcl-2 protein family: Arbiters of cell survival. Science 281:1322–1326.
Altshuler LL, Casanova MF, Goldberg TE, Kleinman JE (1990): The hippocampus and parahippocampus in schizophrenia, suicide, and control brains. Arch Gen Psychiatry 47:1029 – 1034.
Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J (1999): Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett 453:260 –264.
Arendt T, Lehmann K, Seeger G, Gartner U (1999): Synergistic effects of tetrahydroaminoacridine and lithium on cholinergic function after excitotoxic basal forebrain lesions in rat.
Pharmacopsychiatry 32:242–247.
Asghari V, Wang JF, Reiach JS, Young LT (1999): Differential effects of mood stabilizers on Fos/Jun proteins and AP-1 DNA binding activity in human neuroblastoma SH-SY-5Y cells. Mol Brain Res 58:95–102.
Baumann B, Bogerts B (in press): Post-mortem studies on bipolar disorder. Br J Psychiatry.
Baumann B, Danos P, Krell D, Dickmann S, Leschinger A, Stauch R, et al (1999): Reduced volume of limbic system-affiliated basal ganglia in mood disorders: Preliminary data from a postmortem study. J Neuropsychiatry Clin Neurosci 11:71–78.
Benes FM, Kwok EW, Vincent SL, Todtenkopf MS (1998): A reduction of nonpyramidal cells in sector CA2 of schizo-phrenics and manic depressives. Biol Psychiatry 44:88 –97. Bijur GM, De Sarno P, Jope RS (2000): Glycogen synthase
kinase-3bfacilitates staurosporine- and heat shock-induced ap-optosis: Protection by lithium. J Biol Chem 275:7583–7590. Birken DL, Oldendorf WH (1989): N-acetyl-L-aspartic acid: A
literature review of a compound prominent in 1H-NMR spec-troscopic studies of brain. Neurosci Biobehav Rev 13:23–31. Bonfanti L, Strettoi E, Chierzi S, Cenni MC, Liu XH, Martinou
J-C, et al (1996): Protection of retinal ganglion cells from natural and axotomy-induced cell death in transgenic mice overexpressing bcl-2. J Neurosci 16:4186 – 4194.
Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999): Cell survival promoted by the Ras MAPK signaling pathway by transcription-dependent and -indepen-dent mechanisms. Science 286:1358 –1362.
Bruckheimer EM, Cho SH, Sarkiss M, Herrmann J, McDonnell TJ (1998): The Bcl-2 gene family and apoptosis. Adv
Bio-chem Eng Biotechnol 62:75–105.
Bruno V, Sortino MA, Scapagnini U, Nicoletti F, Canonico P (1995): Antidegenerative effects of Mg(21) valproate in cultured cerebellar neurons. Funct Neurol 10:121–30. Cameron HA, McKay RD (1999): Restoring production of
hippocampal neurons in old age. Nat Neurosci 2:894 – 897. Chalecka-Franaszek E, Chuang DM (1999): Lithium activates
the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl
Acad Sci U S A 96:8745– 8750.
Chen DF, Schneider GE, Martinou JC, Tonegawa S (1997): Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 385:434 – 439.
Chen G, Hasanat KA, Bebchuk JM, Moore GJ, Glitz D, Manji HK (1999a): Regulation of signal transduction pathways and gene expression by mood stabilizers and antidepressants.
Psychosom Med 61:599 – 617.
Chen G, Huang LD, Jiang YM, Manji HK (1999b): The mood stabilizing agent valproate inhibits the activity of glycogen synthase kinase 3. J Neurochem 72:1327–1330.
Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK (2000): Enhancement of hippocampal neurogenesis by lith-ium. J Neurochem 75:1729 –1734.
Chen RW, Chuang DM (1999): Long term lithium treatment suppresses p53 and Bax expression but increases bcl-2 expression. J Biol Chem 274:6039 – 6042.
Chen G, Zeng WZ, Jiang L, Yuan PX, Zhao J, Manji HK (1999c): The mood stabilizing agents lithium and valproate robustly increase the expression of the neuroprotective pro-tein bcl-2 in the CNS. J Neurochem 72:879 – 882.
Chuang DM, Wei H, Qin Z, Wei W, Wang Y, Qian Y (1999): Lithium inhibits striatal damage in an animal model of Huntington disease. Soc Neurosci Abstr 241.7.
Dale TC (1998): Signal transduction by the Wnt family of ligands. Biochem J 329:209 –223.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997): Akt phosphorylation BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241.
deBeer R, van den Boogaart A, van Ormondt D, Pijnappel WW, den Hollander JA, Marien AJ, Luyten PR (1992): Application of time-domain fitting in the quantification of in vivo 1H spectroscopic imaging data sets. NMR Biomed 5:171–178. D’Mello SR, Anelli R, Calissano (1994): Lithium induces
apoptosis in immature cerebellar granule cells but promotes survival of mature neurons. Exp Cell Res 211:332–338. Drevets WC (2000): Neuroimaging studies of mood disorders.
Biol Psychiatry 48:813– 828.
Drevets WC, Gadde KM, Krishnan KRR (1999): Neuroimaging studies of mood disorders. In: Charney DS, Nestler EJ, Bunney BS, editors. Neurobiology of Mental Illness. New York: Oxford University Press, 394 – 418.
Duman RS, Heninger GR, Nestler EJ (1997): A molecular and cellular theory of depression. Arch Gen Psychiatry 54:597– 606. English JD, Sweatt JD (1997): A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 272:19103–19106.
Finkbeiner S (2000): CREB couples neurotrophin signals to survival messages. Neuron 25:11–14.
Goldberg JF, Harrow M (1999): Bipolar Disorders: Clinical
Course and Outcome. Washington, DC: American
Psychiat-ric Press.
Goodwin F, Jamison KR (1990): Manic-Depressive Illness. New York: Oxford University Press.
Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ (1999a): Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2:260 –265.
Gould E, Reeves AJ, Graziano MS, Gross CG (1999b): Neuro-genesis in the neocortex of adult primates. Science 286(5439): 548 –552.
Gould E, Tanapat P, Rydel T, Hastings N (2000): Regulation of hippocampal neurogenesis in adulthood. Biol Psychiatry 48: 715–720.
Grignon S, Levy N, Couraud F, Bruguerolle B (1996): Tyrosine kinase inhibitors and cycloheximide inhibit Li1protection of
cerebellar granule neurons switched to non depolarizing medium. Eur J Pharmacol 315:111–114.
Gutkind JS (1998): The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J Biol Chem 273:1839 –1842. Hetman M, Cavanaugh JE, Kimelman D, Xia Z (2000): Role of
glycogen synthase kinase-3beta in neuronal apoptosis in-duced by trophic withdrawal. J Neurosci 20:2567–2574. Hirayasu Y, Shenton ME, Salisbury DF, Dickey CC, Fischer IA,
Mazzoni P, et al (1998): Lower left temporal lobe MRI volumes in patients with first-episode schizophrenia compared with psy-chotic patients with first-episode affective disorder and normal subjects. Am J Psychiatry 155:1384 –1391.
Hong M, Chen DC, Klein PS, Lee VM (1997): Lithium reduces tau phosphorylation by inhibition of glycogen synthase ki-nase-3. J Biol Chem 272:25326 –25332.
Hyman SE, Nestler EJ (1996): Initiation and adaptation: A paradigm for understanding psychotropic drug action. Am J
Psychiatry 153:151–162.
Ikonomov O, Manji HK (1999): Molecular mechanisms under-lying mood-stabilization in manic-depressive illness: The phenotype challenge. Am J Psychiatry 156:1506 –1514. Inouye M, Yamamura H, Nakano A (1995): Lithium delays the
radiation-induced apoptotic process in external granule cells of mouse cerebellum. J Radiat Res (Tokyo) 36:203–208. Joffe RT, Young LT (1997): Bipolar Disorder: Biological
Models and Their Clinical Application. New York: Marcel
Dekker.
Jope RS (1999): Anti-bipolar therapy: Mechanism of action of lithium. Mol Psychiatry 4:117–128.
Kempermann G, Gage FH (1999): Experience-dependent regu-lation of adult hippocampal neurogenesis: Effects of long-term stimulation and stimulus withdrawal. Hippocampus 9:321–332.
Kempermann G, Kuhn HG, Gage FH (1997): More hippocampal neurons in adult mice living in an enriched environment.
Nature 386:493– 495.
Klein PS, Melton DA (1996): A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A 93:8455– 8459.
Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S (1997): Bcl-2: Prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis.
Sci-ence 277:559 –562.
Lenox RH, McNamara RK, Papke RL, Manji HK (1998): Neurobiology of lithium: An update. J Clin Psychiatry 59(suppl 6):37– 47.
Li R, Shen Y, El-Mallakh RS (1994): Lithium protects against ouabain-induced cell death. Lithium 5:211–216.
Liang P, Bauer D, Averboukh L, Warthoe P, Rohrwild M, Muller H, et al (1995): Analysis of altered gene expression by differential display. Methods Enzymol 254:304 –321. Lovestone S, Davis DR, Webster MT, Kaech S, Brion JP, Matus
A, Anderton BH (1999): Lithium reduces tau phosphoryla-tion: Effects in living cells and in neurons at therapeutic concentrations. Biol Psychiatry 45:995–1003.
Lu R, Song L, Jope RS (1999): Lithium attenuates p53 levels in human neuroblastoma SH-SH-SY5Y cells. Neuroreport 10: 1123–1125.
Manji HK, Chen G, Hsiao JK, Masana MI, Moore GJ, Potter WZ (2000a): Regulation of signal transduction pathways by mood stabilizing agents: Implications for the pathophysiology and treatment of bipolar affective disorder. In: Manji HK, Bow-den CL, Belmaker RH, editors. Bipolar Medications:
Mech-anisms of Action. Washington, DC: American Psychiatric
Press, 129 –177.
Manji HK, Moore GJ, Chen G (1999): Lithium at 50: Have the neuroprotective effects of this unique cation been over-looked? Biol Psychiatry 46:929 –940.
Manji HK, Moore GJ, Chen G (2000b): Lithium upregulates the cytoprotective protein bcl-2 in vitro and in the CNS in vivo: A role for neurotrophic and neuroprotective effects in manic-depressive illness. J Clin Psychiatry 61(suppl 9):82–96. Manji HK, Potter WZ, Lenox RH (1995): Signal transduction
pathways: Molecular targets for lithium’s action. Arch Gen
Psychiatry 52:531–543.
Mark RJ, Ashford JW, Goodman Y, Mattson MP (1995): Anticonvulsants attenuate amyloid beta peptide neurotoxic-ity, Ca21 deregulation, and cytoskeletal pathology.
Neuro-biol Aging 16:187–198.
Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER (1997): MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilita-tion in Aplysia. Neuron 18:899 –912.
Mayberg HS, Brannan S, Tekell JL, Silva JA, Mahurin RK, McGinnis S, Jerabek P (2000): Regional metabolic effects of fluoxetine in major depression: Serial changes and relation-ship to clinical response. Biol Psychiatry 48:830 – 843. Merry DE, Korsmeyer SJ (1997): Bcl-2 gene family in the
nervous system. Annu Rev Neurosci 20:245–267.
Moore GJ, Bebchuk JM, Hasanat K, Chen G, Seraji-Bozorgzad N, Wilds IB, et al (2000): Lithium increases N-acetyl-aspartate in the human brain: In vivo evidence in support of bcl-2’s neurotrophic effects? Biol Psychiatry 48:1– 8. Moore GJ, Bebchuk JM, Parrish JK, Faulk MW, Arfken CL,
Moore GJ, Bebchuk J, Wilds IB, Chen G, Manji HK (in press): Pharmacologic increase in human brain grey matter. Lancet. Mora A, Gonzalez-Polo RA, Fuentes JM, Soler G, Centeno F (1999): Different mechanisms of protection against apoptosis by valproate and Li1. Eur J Biochem 266:886 – 891. Munoz-Montano JR, Moreno FJ, Avila J, Diaz-Nido J (1997):
Lithium inhibits Alzheimer’s disease-like tau protein phos-phorylation in neurons. FEBS Lett 411:183–188.
Murray CJ, Lopez AD (1997a): Alternative projections of mortality and disability by cause 1990 –2020: Global Burden of Disease Study. Lancet 349:1498 –1504.
Murray CJ, Lopez AD (1997b): Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 349:1436 –1442.
Nestler EJ (1998): Antidepressant treatments in the 21st century.
Biol Psychiatry 44:526 –533.
Nishimura M, Yu G, Levesque G, Zhang DM, Ruel L, Chen F, et al (1999): Presenilin mutations associated with Alzheimer disease cause defective intracellular trafficking of beta-cate-nin, a component of the presenilin protein complex. Nat Med 5:164 –169.
Nonaka S, Chuang DM (1998): Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats.
Neurore-port 9:2081–2084.
Nonaka S, Hough CJ, Chuang DM (1998a): Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-as-partate receptor-mediated calcium influx. Proc Natl Acad Sci
U S A 95:2642–2647.
Nonaka S, Katsube N, Chuang DM (1998b): Lithium protects rat cerebellar granule cells against apoptosis induced by anticon-vulsants, phenytoin and carbamazepine. J Pharmacol Exp
Ther 286:539 –547.
Ozaki N, Chuang DM (1997): Lithium increases transcription factor binding to AP-1 and cyclic AMP-responsive element in cultured neurons and rat brain. J Neurochem 69:2336 –2344. Pap M, Cooper GM (1998): Role of glycogen synthase kinase-3 in the phosphatidylinositol 3 Kinase/Akt cell survival path-way. J Biol Chem 273:19929 –19932.
Pascual T, Gonzalez JL (1995): A protective effect of lithium on rat behavior altered by ibotenic acid lesions of the basal forebrain cholinergic system. Brain Res 695:289 –292. Rajkowska G (1997): Morphometric methods for studying the
prefrontal cortex in suicide victims and psychiatric patients.
Ann N Y Acad Sci 836:253–268.
Rajkowska G (2000): Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol
Psychiatry 48:766 –777.
Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al (1999): Morphometric evidence for neuro-nal and glial prefrontal cell pathology in major depression.
Biol Psychiatry 45:1085–1098.
Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD (1999): Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358 –2361.
Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C,
Sweatt JD (1999): The mitogen activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocam-pus. J Neurosci 19:4337– 4348.
Segal RA, Greenberg ME (1996): Intracellular signaling path-ways activated by neurotrophic factors. Annu Rev Neurosci 19:463– 489.
Sheline YI (2000): 3-D MRI studies of neuroanatomic changes in unipolar major depression: The role of stress and medical comorbidity. Biol Psychiatry 48:791– 800.
Smith MA, Makino S, Altemus M, Michelson D, Hong SK, Kvetnansky R, Post RM (1995): Stress and antidepressants differentially regulate neurotrophin 3 mRNA expression in the locus Coeruleus. Proc Natl Acad Sci U S A 92:8788 – 8792.
Sparapani M, Virgili M, Ortali F, Contestabile A (1997): Effects of chronic lithium treatment on ornithine decarboxylase induction and excitotoxic neuropathology in the rat. Brain
Res 765:164 –168.
Strakowski SM, Wilson DR, Tohen M, Woods BT, Douglass AW, Stoll AL (1993): Structural brain abnormalities in first-episode mania. Biol Psychiatry 33:602– 609.
Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998): Ca21influx regulates BDNF transcription by a CREB
family transcription factor-dependent mechanism. Neuron 20:709 –726.
Tsai G, Coyle JT (1995): N-Acetylaspartate in neuropsychiatric disorders. Prog Neurology 46:531–540.
van den Boogaart A, Ala-Korpela M, Jokisaari J, Griffiths JR (1994): Time and frequency domain analysis of NMR data compared: An application to 1D 1H spectra of lipoproteins.
Magn Reson Med 31:347–358.
van Praag H, Kempermann G, Gage FH (1999): Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2:266 –270.
Vincent SL, Todtenkopf MS, Benes FM (1997): A comparison of the density of pyramidal and nonpyramidal neurons in the anterior cingulate cortex of schizophrenics and manic depres-sives. Soc Neurosci 23:2199.
Volonte C, Rukenstein A (1993): Lithium promotes short-term survival of PC12 cells after serum and NGF deprivation.
Lithium 4:211–219.
Wang JF, Bown C, Young LT (1999): Differential display PCR reveals novel targets for the mood-stabilizing drug valproate including the molecular chaperone GRP78. Mol Pharmacol 55:521–527.
Wang JF, Young LT (1996): Differential display PCR reveals increased expression of 29,39-cyclic nucleotide 39 -phosphodi-esterase by lithium. FEBS Lett 386:225–229.
Willert K, Nusse R (1998): Beta-catenin: A key mediator of Wnt signaling. Curr Opin Genet Dev 8:95–102.
Yuan PX, Chen G, Manji HK (1999): Lithium activates the c-Jun NH2-terminal kinases (JNKs) in vitro and in the CNS in vivo.
J Neurochem 73:2299 –2309.