www.elsevier.com / locate / bres
Research report
Hyperglycemic but not normoglycemic global ischemia induces
marked early intraneuronal expression of
b
-amyloid precursor protein
*
Baowan Lin, Myron D. Ginsberg , Raul Busto
Cerebral Vascular Disease Research Center, Department of Neurology, University of Miami School of Medicine, Miami, FL 33101, USA
Accepted 19 September 2000
Abstract
Preischemic hyperglycemia is known to accentuate acute ischemic injury to neurons, microglia, and endothelia. In the present study, we used a monoclonal antibody to the N-terminal portion of b-APP to examine how the immunoreactivity of this normal membrane glycoprotein is differentially influenced by transient cerebral ischemia when carried out under normoglycemic vs. hyperglycemic conditions. Anesthetized, physiologically regulated rats received 12.5 min of global forebrain ischemia by bilateral carotid artery occlusions plus systemic hypotension. Hyperglycemia was induced by intraperitoneal dextrose administration prior to ischemia. One or three days later, brains were examined byb-APP immunohistochemistry. Ischemia under hyperglycemic conditions led to the robust, widespread intraneuronal expression ofb-APP immunoreactivity in neocortex, hippocampus, thalamus, and striatum of all 11 rats; this was most prominent at 24 h postischemia. Compared to rats with normoglycemic ischemia, numbers ofb-APP-immunopositive neurons in the parietal cortex of hyperglycemic rats were increased by 5.9 fold at 24 h, and by 10.6 fold at 3 days postischemia.
b-APP-immunopositive neurons in hyperglycemic rats often exhibited striking morphological alterations typical of ischemic necrosis; however, no b-APP immunoreaction was observed in zones of frank infarction. Brains of normoglycemic rats (n511), by contrast, showed only weakb-APP immunostaining in occasional non-necrotic pyramidal neurons of parietal neocortex; no necrosis was present in thalamus. In sham-operated hyperglycemic rats, b-APP immunostaining of thalamic neurons was somewhat increased at 24 h. Western analysis revealed that the hyperglycemia-induced intraneuronal overexpression of b-APP was not associated with an overall increase in tissue levels. The results of this study demonstrate that transient forebrain ischemia under hyperglycemic conditions leads to the early intraneuronal expression of b-APP within neuronal populations showing a heightened susceptibility to hyperglycemia-induced accentuation of ischemic injury. Our data suggest thatb-APP or its metabolites may be involved in the injury process. 2001 Elsevier Science B.V. All rights reserved.
Keywords: Hyperglycemia; Amyloid; Selective vulnerability; Immunochemistry; Rat
1. Introduction determine its physiological or pathological role. Increased
b-APP levels have been noted in acute cerebral ischemia Acute hyperglycemia markedly accentuates the neuro- [28,29,33,39,41], and we have shown thatb-APP accumu-pathological alterations resulting from cerebral ischemia lates, as well, in the subacute and chronic stages after a [7,10,16,24,31], but the mechanisms underlying this effect brief episode of global forebrain ischemia in nor-are not fully understood.b-amyloid precursor protein (b- moglycemic rats [18]. However, the influence of hy-APP) comprises a group of highly conserved 100–140 kDa perglycemia on ischemia-induced changes of b-APP has integral membrane glycoproteins expressed in almost all not been studied. In this immunohistochemical study, we mammalian cells and present in normal neuronal perikarya, employed a monoclonal antibody to the N-terminal region proximal dendrites and axons [20,22,37] as well as in of b-APP [18] to explore whether hyperglycemia influ-blood vessels, meningeal membranes, and ependyma [6]. ences the postischemic expression of this important
pro-b-APP is metabolized via several alternative pathways that tein.
*Corresponding author. Tel.: 11-305-243-6449; fax: 11-305-243- 2. Materials and methods 5830.
E-mail address: [email protected] (M.D. Ginsberg). These studies were conducted in male Wistar rats
weighing 300–350 g following an overnight fast. The n56) or 3 days (n55 in each group) after the ischemic University of Miami’s Animal Care and Use Committee insult. Ten sham animals were similarly studied (hy-approved all procedures. Anesthesia was induced with 3% perglycemic, 24 h survival, n54; hyperglycemic, 3 day halothane and 70% nitrous oxide. Animals were intubated survival, n53; normoglycemic, 3 day survival, n53). Rats endotracheally and ventilated mechanically on mixtures of were reanesthetized by halothane and perfused via the 0.5% halothane, 70% nitrous oxide and a balance of ascending aorta with FAM (a mixture of 40% formalde-oxygen. The femoral arteries were catheterized for blood hyde, glacial acetic acid and methanol, 1:1:8 by volume) pressure monitoring and to permit arterial sampling for for 19 min at a pressure of 100–120 mmHg following a 1 blood–gas measurements. Arterial PCO2 and PO2 were min perfusion with physiological saline. The heads were maintained in the normal range by ventilatory adjustments. immersed in FAM at 48C for 1 day. The brain was then Rats were immobilized by pancuronium bromide, 0.75 removed, placed in FAM for another day, and then mg / kg i.v. Rectal temperature was measured continually blocked. Coronal tissue blocks were embedded in paraffin and maintained at 37.0–37.58C by a warming lamp above for sectioning. Brain sections (10mm thick) were prepared the rat’s body. Cranial temperature was separately moni- at 250 mm intervals and were stained for the light tored by a 29-gauge thermocouple implanted into the left microscopic immunocytochemical visualization of the N-temporalis muscle and was maintained at 36.0–36.58C terminal portion ofb-APP(695) (clone 22C11, 0.1mg / ml, throughout the experiment by a small warming lamp above Boehringer Mannheim).
the rat’s head. (In our previous studies, we have shown For b-APP immunohistochemistry, sections were rehy-that this corresponds to a brain temperature of 36.5–37.08C drated and placed in 1.5% H O2 2 in methanol to block
[5].) endogenous peroxidase activity. They were then rinsed in
running water, dipped in 0.05 M phosphate-buffered saline (PBS), and incubated with normal horse serum. The
2.1. Hyperglycemia primary antibody (1:500) was applied at 48C for 18 h. To
test for nonspecific staining, negative controls were con-To induce hyperglycemia, rats received an intraperi- ducted in which the primary antibody was omitted and, toneal injection of 2.5 ml of 25% dextrose solution 30 min instead, mouse IgG1 (1:500) was used (No. X0931, Dako prior to initiating the ischemic insult. Normoglycemic rats Corp., Carpenteria, CA). Sections then were rinsed with were given 2.5 ml of sterile water i.p. PBS and the secondary antibody applied. The avidin– biotin complex (Vector, Burlingame, CA) was used for 2.2. Global forebrain ischemia antibody detection followed by peroxidase reaction with diaminobenzidine (DAB) and H O . Slides were washed2 2
Both common carotid arteries were exposed via a in 0.5% Triton X-100 with or without counterstaining by midline ventral incision and were gently separated from hematoxylin. These procedures were similar to those the surrounding nerve fibers. Ligatures consisting of recently reported from our laboratory [18].
polyethylene PE-10 tubing contained within a double-lumen Silastic tubing were passed loosely around each
artery. To produce ischemia, arterial blood was gradually 2.4. Identification and quantitation ofb-APP withdrawn into a heparanized syringe to reduce mean immunoreactivity
arterial blood pressure (MAP) to 45 mmHg. Global
ischemia was then produced by tightening the carotid Representative areas of neocortex, striatum (coronal ligatures bilaterally and maintaining blood pressure at 45 level, 0.2 mm posterior to bregma), and the hippocampal mmHg for 12.5 min [17]. The ischemic insult was ended CA1 sector and thalamus (3.6 mm posterior to bregma) by removing the carotid ligatures and immediately reinfus- were examined by an observer (B.L.) blinded to the ing the shed blood (which had been maintained at body experimental groups. Specific b-APP staining was iden-temperature) to restore MAP to 100–120 mmHg. Catheters tified by its dark brown appearance, which often exhibited were then removed, incisions closed, and rats were re- a fine granular punctate pattern. All immunohistochemical turned to their cages and given free access to food and and histological findings were carefully confirmed by a
water. second observer (M.D.G.). Counting of b-APP-positive
Sham-operated rats received similar operative prepara- cells was performed in a blinded fashion within a stan-tion but were not subjected to carotid occlusion or blood dardized 103 microscopic field of the lateral parietal
withdrawal. neocortex of each hemisphere (region Par2 [27], coronal
level of 3.6 mm posterior to bregma). Left- and right-2.3. Tissue preparation and immunohistochemistry hemisphere counts were summed for each rat.
2.5. Quantitative western blots 2.6 fold by dextrose administration (hyperglycemic–is-chemic rats, 340666 mg / dL; normoglycemic–ischemic In a separate series, relative levels of expression of group, 133621 mg / dl).
b-APP were determined by quantitative western blot
analysis. The following experimental groups were studied: 3.2. Summary of light-microscopic histopathology sham / normoglycemic (n53) and sham / hyperglycemic
(n53) with 24 h recovery; 12.5 min ischemia 124 h The light-microscopic neuropathological alterations in recovery / normoglycemic (n54), 12.5 min ischemia124 h normoglycemic and hyperglycemic rats surviving for 1 and recovery / hyperglycemic (n54); 12.5 min ischemia 13 3 days following a 12.5 min ischemic insult have been day recovery / normoglycemic (n54); and 12.5 min is- previously reported and illustrated in detail [16]. To chemia 13 day recovery / hyperglycemic (n54). Tissue summarize these findings, brains of hyperglycemic –is-samples from hippocampus (|100 mg), thalamus (|40 chemic rats contained extensive ischemic neuronal altera-mg), and cortex (|500 mg) of each brain were isolated and tions and foci of infarction within neocortex, striatum, and rapidly frozen on dry-ice. Total protein was isolated by thalamus; and widespread hippocampal damage extending homogenization in buffer composed of 50 mM Tris, pH beyond the CA1 sector. These changes were already 8.0, 100 mM NaCl, 0.1% SDS, 0.01 mg / ml leupeptin, and apparent at 24 h. Vascular changes observed in the hy-100 mM phenylmethylsulfonyl fluoride (PMSF). Protein perglycemic group consisted of endothelial thickening, concentration was determined using the Bio-Rad DC vascular occlusion, prominent peri- and intravascular
poly-protein assay system. morphonuclear and monocytic accumulation, and foci of
Protein samples were added to 10% SDS and boiled to perivascular rarefaction and microinfarction. By contrast, denature them. Twenty five mg of protein per sample was normoglycemic–ischemic rats showed no injury at 24 h. loaded onto a 12% SDS polyacrylamide stacking gel and At 3 days, the cerebral neocortex and striatum of nor-electrophoresed in 25 mM Tris, 250 mM glycine pH 8.3, moglycemic–ischemic animals showed only mild damage, 0.1% SDS at 30 mA. Proteins were transferred by standard with few or no necrotic neurons; the thalamus was spared electroblotting techniques to a PVDF transfer membrane from injury; hippocampal damage was confined to the CA1 (Polyscreen, NEN Research Products) in 25 mM Tris, 192 sector; and no endothelial changes were present. Numbers mM glycine, pH 8.3, and 10% methanol. After confirma- of ischemic cells in the striatum of hyperglycemic animals tion of transfer by Ponceau S Red staining, the membrane were increased by 5 fold or more compared to nor-was blocked with 5% milk in 13PBS plus Tween-20 for 2 moglycemic rats, and surviving (normal) neurons in the
h at room temperature. vulnerable hippocampal CA1 sector of hyperglycemic–
Incubation with a monoclonal antibody to the N-termi- ischemic rats were reduced to one third of the number seen nal portion of b-APP(695) (clone 22C11, 0.1 mg / ml, in normoglycemic animals [16].
Boehringer Mannheim) was performed in the same buffer
at 1:1000 dilution at room temperature for 2 h. Following a 3.3. Immunohistochemical observations brief wash, incubation with HRP-linked anti-mouse IgG
(1:1000) was carried out for 45 min. Detection of signal 3.3.1. Sham-operated group
was performed with a Phototope-HRP Western blot de- In four of six sham brains (3 normoglycemic, 1 hy-tection kit (New England BioLabs) utilizing LumiGlo perglycemic) studied at 3 days, scattered faintly stained, reagent and was visualized on X-ray film. The film was apparently extracellular foci of b-APP immunoreactivity then digitized, and densitometric measurements were made were observed on non-counterstained sections, randomly to quantify signals from each sample. Following stripping distributed within the middle layers of neocortex. These of the antibody, the blot was re-incubated as above with an foci, however, lacked both a dark brown appearance and a antibody against actin (Sigma Chemicals) at 1:5000. The punctate or granular texture in hematoxylin-counterstained actin signal was used as an internal control for evenness of sections and were only rarely intraneuronal. In three of
loading. four hyperglycemic sham-operated rats with 24 h survival,
widespread thalamic neurons tended to exhibit light brown cytoplasmic b-APP immunostaining that was not present 3. Results in the other sham-operated groups. This faint staining clearly differed, however, from that to be described below 3.1. Physiological variables in hyperglycemic–ischemic brains, and it was not observed in neocortex or hippocampus sites prominently affected by Mean blood pressure before ischemia averaged 125610 hyperglycemic ischemia (see below).
mmHg (mean6S.D.); arterial pO2 was 117622 mmHg;
arterial pCO , 402 63 mmHg; and arterial pH, 7.4260.02. 3.3.2. Hyperglycemic–ischemic brains — 24 h
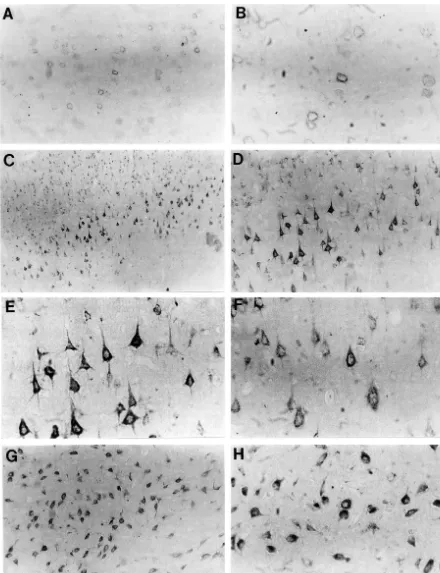
immuno-reactivity involving neurons throughout the neocortex (Fig. immunoreactivity was observed within shrunken pyramidal 1C–F), hippocampus (Fig. 2), and dorsal and ventrolateral neurons of frontoparietal neocortical layer V (Fig. 3) thalamus (Fig. 1G,H). In neocortex, robust intraneuronal (although not in regions of frank infarction). Fewer
num-b-APP immunostaining consistently involved neurons of bers of neurons were affected than at 24 h, however, and the cingulate gyrus and of all layers of dorsolateral and neurons of cingulate gyrus were typically uninvolved. All lateral neocortex; large pyramidal neurons were the most four brains with cortical b-APP immunopositivity also consistently affected (Fig. 1). On hematoxylin-counter- contained prominent b-APP reactivity throughout the stained sections, b-APP-positive neocortical neurons were, entire pyramidal layer of the hippocampal CA1 sector and in some cases, shrunken and triangular in shape with subiculum;b-APP-immunopositive neurons were shrunken altered nuclear staining, suggesting ischemic necrosis. and appeared necrotic. By contrast, the (non-necrotic) Otherb-APP-positive cortical neurons, however, were not neurons of the CA3 sector were immunonegative. One shrunken, showed normal nuclear and nucleolar morpholo- brain displayed CA1 necrosis and b-APP immuno-gy, and appeared to be viable. Adjacent H&E-stained positivity on one side, but normal CA1 morphology and sections confirmed that cortical zones of b-APP-immuno- b-APP immunonegativity on the other. The thalamus positivity corresponded to foci of extensive neuronal contained b-APP-positive neurons in only two cases; and necrosis, with shrunken cellular contours, cytoplasmic scattered small cells of the striatum wereb-APP-positive eosinophilia, and nuclear condensation. No b-APP-im- in three cases.
munoreactivity was noted, however, in areas of frank
infarction. 3.3.5. Normoglycemic–ischemic brains — 3 days
In the hippocampus of 24 h hyperglycemic–ischemic Weak intraneuronal b-APP immunostaining was ob-rats (Fig. 2), b-APP-immunostained sections of all six served only in occasional neurons of parietal neocortex in brains revealed prominent b-APP-positivity involving all four of five brains studied at 3 days. No b-APP-reactivity hippocampal sectors and extending into the dentate hilus was present in thalamus, striatum, or hippocampus. (Fig. 2E,F). On H&E sections, the hippocampal CA1 No extracellularb-APP immunoreactivity was observed sector of these brains contained extensive bilateral is- in any ischemic brain.
chemic necrosis of pyramidal neurons in two of six brains;
predominantly unilateral alterations in two brains; and an 3.4. Quantitation of neocortical b-APP immunoreactivity absence of ischemic neuronal changes in the remaining
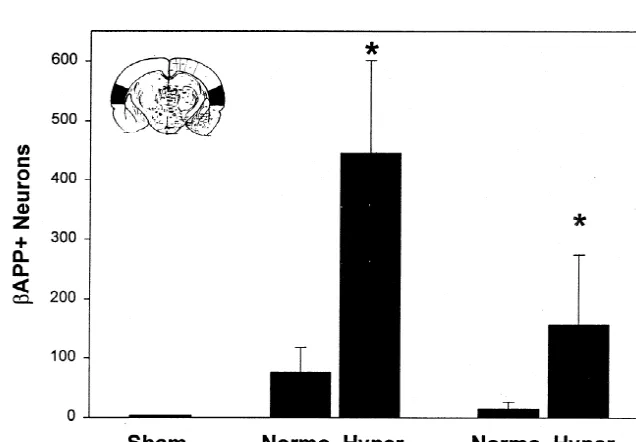
two cases. In all brains of this group, the dorsal and Quantitative analysis of b-APP-positive neurons in ventrolateral regions of thalamus also contained extensive parietal neocortex of hyperglycemic–ischemic rats re-zones of markedly b-APP-positive neurons (Fig. 1G,H); vealed 5.9 fold mean elevations at 24 h, and 10.6 fold adjacent H&E sections revealed these neurons to have the elevations at 3 days, compared to counts in animals with classic morphologic features of ischemic necrosis. Five of normoglycemic ischemia (Fig. 4). These differences were six rats at 24 h exhibited mild intraneuronal b-APP highly significant.
reactivity within small cells of the striatum. No b-APP
immunoreactivity was present in the corpus callosum. 3.5. Western analysis
3.3.3. Normoglycemic–ischemic brains — 24 h Western blots in all groups revealed a single consistent The six animals of this group showed only low-density band (data not shown). By quantitative densitometry, the intraneuronal b-APP immunostaining of larger pyramidal overall tissue levels ofb-APP in cortex, hippocampus, and neurons within the dorsolateral and lateral regions of thalamus were similar in normoglycemic- and hy-frontoparietal neocortex (Fig. 1A,B). On hematoxylin- perglycemic–ischemic rats and in sham animals. These counterstained sections, this b-APP immunopositivity in- data, normalized to actin, are shown in Table 1 for volved the cytoplasm but not the nuclear region of neurons hippocampus and thalamus. (Actin-normalization was not and was punctate or granular in texture. b-APP-positive available for cortical samples, but no intergroup differ-neurons appeared morphologically normal, with intact ences in background-corrected optical densities forb-APP nuclear morphology, visible nucleolus and no cellular were evident.)
shrinkage. The cingulate gyrus and thalamus contained no
b-APP immunopositivity. The CA1 sector of hippocampus
showed faint b-APP immunoreaction within normal ap- 4. Discussion pearing neurons in two of six brains; and scattered large
neurons of the dentate hilus exhibited consistent b-APP Our results clearly demonstrate for the first time that, in
immunostaining. contrast to the normoglycemic–ischemic condition,
Fig. 2.b-APP immunohistochemistry of hippocampus 24 h after normoglycemic (A) or hyperglycemic (B–F) ischemia. Panels A–D: CA1 sector. Panels E, F: dentate hilus. Normoglycemic–ischemic brain (A) shows nob-APP immunoreactivity in CA1 sector. By contrast, hyperglycemic ischemia (B–D) leads to intenseb-APP immunopositivity throughout CA1 sector. Region designated by arrowhead (Panel B) is shown at higher power in Panel C. Some b-APP immunopositive CA1 neurons appear morphologically intact (D) although by H&E stain they exhibit eosinophilia. Other subfields of CA1 (C) exhibit clear evidence of typical ischemic necrosis. Dentate hilar neurons also show striking b-APP immunopositivity (E, F) (affected neurons were eosinophilic on H&E stain). (Magnifications: A:3400; B, E:3200; C, D, F:3800).
involvement of neurons of neocortex, the entire hippocam- acuteb-APP immunoreactivity in the normoglycemic rats pus, dorsal and ventrolateral regions of thalamus, and of the present study resembles that reported by other small cells of the striatum. This prominent intraneuronal authors [29,39,41,43]. It is highly unlikely that the
Fig. 3. Frontoparietal neocortex, three day survival: Panel A: Normoglycemic–ischemic brain shows mildb-APP immunostaining of occasional cortical neurons. Panels B–D: hyperglycemic ischemia. Widespread intraneuronalb-APP immunoreactivity is present within large numbers of shrunken pyramidal neurons that, on H&E stains, appear necrotic. (Magnifications: A, C, D:3800; B:3400).
Table 1
Data from rats with 12.5 min of global forebrain ischemia followed by recirculation for 24 h or 3 days; and from sham-ischemic rats with 24 h survival. Values represent optical densities (means6S.E.M.), background-corrected and normalized to respective OD values for actin.
cresyl violet stains; and (2) b-APP reactivity was absent the repair of vascular injury and to cell growth within frankly infarcted regions. Western analysis of [9,21,22,34,36]. When the secreted form of b-APP is regional brain samples weighing |40–500 mg revealed administered intracerebroventricularly after ischemia in that the prominent specifically intraneuronal expression of rats, hippocampal CA1 neurons are protected against
b-APP in hyperglycemic–ischemic neurons was not re- ischemic injury [38]. On the other hand, other evidence flected in an overall increase in tissue levels of this supports the possibility that b-APP may also exert a constitutively expressed protein. Rather, these data, taken neurotoxic effect. The copper-binding domain of b-APP together, suggest either that hyperglycemic ischemia en- mediates the reduction of copper (II) to copper (I) [23]. abled the redistribution of b-APP into the intraneuronal Excessive copper (I) and hydroxyl radical formation can compartment; or that b-APP was overexpressed within induce oxidative stress in neurons [11,23]. In a recent neurons but that these highly localized increases were study [44], neuronal cultures derived from wild-type mice diluted by the large tissue samples used for western were shown to be significantly more susceptible to toxicity
analysis. induced by physiological concentrations of copper than
The immunohistochemical visualization of expressed were cultures derived fromb-APP-knockout (APP 2/2) proteins depends upon the precise assay conditions em- mice. This was supported by the finding of significantly ployed. b-APP is a constitutive glycoprotein that is higher levels of copper-induced lipid peroxidation in wild-normally present throughout the nervous system type neurons, and by the potentiation of copper toxicity by [6,20,22,37]. (Our western analysis confirmed this fact.) In treating neuronal cultures with a peptide containing the the present study, we avoided the use of cupric sulfate, APP copper-binding domain [44].
employed by other workers [8] to enhance otherwise weak b-APP is transported by fast axonal flow and accumu-immunochemical reactivity. Under these experimental lates in injured CNS axons in relation to ischemic lesions conditions, constitutiveb-APP expression in sham animals [8,25,40,46]. Previous studies of global forebrain ischemia was generally below the threshold of immunohistochemi- have noted increases in b-APP in both vulnerable cal detection, and patterns of b-APP immunoreactivity [12,14,29] and non-vulnerable brain regions [42,43]. Ultra-resulting from hyperglycemic vs. normoglycemic ischemia structural examination of neurons with intense b-APP could be readily differentiated. immunoreactivity has disclosed damage resembling that By comparing the foci of prominent intraneuronal b- seen in central chromatolysis [41]. At three or more days APP immunoreactivity to adjacent sections stained with following 30 min global ischemia, strongb-APP immuno-H&E for conventional microscopic analysis, we were able reactivity has been reported in both astrocytes and mi-to establish that b-APP-positive neurons of hy- croglia in vulnerable areas [1].
perglycemic–ishemic brains correspond closely to regions In focal cerebral ischemia, the delayed induction of showing the classical features of ischemic neuronal necro- APP770 and APP751 mRNA has been noted within the sis. These observations thus suggest that the intraneuronal infarct core and a thin perifocal zone [15]; and b-APP expression of b-APP immunoreactivity following hy- immunoreactivity has been described within axonal swell-perglycemic ischemia is an early marker of enhanced ings, dystrophic neurites, neuronal parikarya [39] and injury. Consistent with this idea is our finding of robust astrocytic processes [13] at the infarct periphery, and in intraneuronalb-APP accumulation in the thalamus at 24 h macrophages [30]. When MCA occlusion was produced in after hyperglycemia ischemia, as this region ordinarily transgenic mice overexpressing b-APP (APP695), the exhibits no ischemic changes following 12.5 min of resulting infarct was enlarged by one third over that in normoglycemic ischemia but is injured by hyperglycemic nontransgenic littermates; this was attributed, however, to
ischemia [16]. a more severe degree of local ischemia in the transgenics
Previous studies have ascribed both beneficial and [47].
for in vivo functions and therapeutic applications, Adv. Biochem. elimination is thought to be a critical determinant of
Eng. Biotechnol. 64 (1999) 155–201. normal neuronal function [19]. A large body of evidence
[4] M. Brownlee, The pathological implications of protein glycation, links the hyperglycemia of diabetes mellitus both with Clin. Invest. Med. 18 (1995) 275–281.
enhanced enzymatic glycosylation [2] and with the acceler- [5] R. Busto, W.D. Dietrich, M.Y. Globus, I. Valdes, P. Scheinberg, M.D. ated formation of irreversible nonenzymatic advanced Ginsberg, Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury, J. Cereb. glycosylation end products (AGEs) that may accumulate in
Blood Flow Metab. 7 (1987) 729–738. the vessel wall and other sites and contribute to the
[6] F. Coria, A. Moreno, A. Torres, I. Ahmad, J. Ghiso, Distribution of complications of chronic diabetes [4]. High glucose con- Alzheimer’s disease amyloid protein precursor in normal human and centrations, putatively acting via the glycosylation of rat nervous system, Neuropathol. Appl. Neurobiol. 18 (1992) 27– nascent proteins, have been shown to impair excitation– 35.
[7] G. de Courten-Myers, R.E. Myers, L. Schoolfield, Hyperglycemia contraction in cardiac myocytes [32]. By analogy, it is
enlarges infarct size in cerebrovascular occlusion in cats, Stroke 19 possible that pre-ischemic hyperglycemia might change the
(1988) 623–630. glycosylation state of b-APP and, thereby, alter its
sub-[8] W.D. Dietrich, S. Kraydieh, R. Prado, N.E. Stagliano, White matter sequent processing and affect neuronal function, although alterations following thromboembolic stroke: a beta-amyloid pre-this conjecture is as yet unproven. The oxidation of cursor protein immunocytochemical study in rats, Acta Neuropathol.
(Berl.) 95 (1998) 524–531. glycated proteins and the interaction of AGEs with
cell-[9] K. Furukawa, S.W. Barger, E.M. Blalock, M.P. Mattson, Activation surface receptors produces superoxide radicals and,
pos-of K1channels and suppression of neuronal activity by secreted sibly, reactive nitrogen species, that are thought to mediate
beta-amyloid-precursor protein, Nature 379 (1996) 74–78. the cellular toxicity of heavily glycated proteins [19,45]. [10] M.D. Ginsberg, F.A. Welsh, W.W. Budd, Deleterious effect of Although protein glycosylation has been intensively glucose pretreatment on recovery from diffuse cerebral ischemia in the cat. I. Local cerebral blood flow and glucose utilization, Stroke studied (see [3,35] for recent reviews), the role of acute
11 (1980) 347–354. hyperglycemia in the glycosylation of neural proteins, and
[11] M.R. Gunther, P.M. Hanna, R.P. Mason, M.S. Cohen, Hydroxyl its pathophysiological implications, remain to be
eluci-radical formation from cuprous ion and hydrogen peroxide: a
spin-dated. trapping study, Arch. Biochem. Biophys. 316 (1995) 515–522.
In summary, our results demonstrate that a brief episode [12] C. Heurteaux, V. Bertaina, C. Widmann, M. Lazdunski, K1channel of forebrain ischemia, when carried out in hyperglycemic openers prevent global ischemia-induced expression of c-fos, c-jun, heat shock protein, and amyloid beta-protein precursor genes and animals, triggers the early robust expression of b-APP
neuronal death in rat hippocampus, Proc. Natl. Acad. Sci. USA 90 within neurons of neocortex, hippocampus, thalamus and
(1993) 9431–9435.
striatum. Our data suggest that the immunohistochemical [13] R.N. Kalaria, S.U. Bhatti, E.A. Palatinsky, D.H. Pennington, E.R. appearance of b-APP within vulnerable neurons is a Shelton, H.W. Chan, G. Perry, W.D. Lust, Accumulation of the beta marker of their heightened susceptibility to hy- amyloid precursor protein at sites of ischemic injury in rat brain,
Neuroreport 4 (1993) 211–214. perglycemic–ischemic injury.
[14] K. Kogure, H. Kato, Altered gene expression in cerebral ischemia, Stroke 24 (1993) 2121–2127.
[15] J. Koistinaho, I. Pyykonen, R. Keinanen, T. Hokfelt, Expression of beta-amyloid precursor protein mRNAs following transient focal Acknowledgements ischaemia, Neuroreport 7 (1996) 2727–2731.
[16] B. Lin, M.D. Ginsberg, R. Busto, Hyperglycemic exacerbation of This study was supported by NIH Grant NS05820 neuronal damage following forebrain ischemia: microglial, as-trocytic and endothelial alterations, Acta Neuropathol. (Berl.) 96 (Research Center for Cerebral Vascular Disease). Mr.
(1998) 610–620. Guillermo Fernandez, Ms. Isabel Saul, Ms. Lin Li, and Ms.
[17] B. Lin, M.D. Ginsberg, R. Busto, W.D. Dietrich, Sequential analysis Jessie Truettner contributed valuable technical assistance. of subacute and chronic neuronal, astrocytic and microglial altera-The authors are grateful to Dr. Rainald Schmidt-Kastner tions after transient global ischemia in rats, Acta Neuropathol. for carefully reading the manuscript and offering critical (Berl.) 95 (1998) 511–523.
[18] B. Lin, R. Schmidt-Kastner, R. Busto, M.D. Ginsberg, Progressive suggestions.
parenchymal deposition of beta-amyloid precursor protein in rat brain following global cerebral ischemia, Acta Neuropathol. (Berl.) 97 (1999) 359–368.
[19] A.W. Lyckman, A.M. Confaloni, G. Thinakaran, S.S. Sisodia, K.L. References Moya, Post-translational processing and turnover kinetics of pre-synaptically targeted amyloid precursor superfamily proteins in the [1] R.B. Banati, J. Gehrmann, C. Wiessner, K.A. Hossmann, G.W. central nervous system, J. Biol. Chem. 273 (1998) 11100–11106.
Kreutzberg, Glial expression of the beta-amyloid precursor protein [20] L.J. Martin, S.S. Sisodia, E.H. Koo, L.C. Cork, T.L. Dellovade, A. (APP) in global ischemia, J. Cereb. Blood Flow Metab. 15 (1995) Weidemann, K. Beyreuther, C. Masters, D.L. Price, Amyloid 647–654. precursor protein in aged nonhuman primates, Proc. Natl. Acad. Sci. [2] H. Bar-On, G. Nesher, A. Teitelbaum, E. Ziv, Dolichol-mediated USA 88 (1991) 1461–1465.
[22] M.P. Mattson, Cellular actions of beta-amyloid precursor protein and [35] P. Sears, C.H. Wong, Enzyme action in glycoprotein synthesis, Cell its soluble and fibrillogenic derivatives, Physiol. Rev. 77 (1997) Mol. Life Sci. 54 (1998) 223–252.
1081–1132. [36] D.J. Selkoe, Alzheimer’s disease: a central role for amyloid, J. [23] G. Multhaup, A. Schlicksupp, L. Hesse, D. Beher, T. Ruppert, C.L. Neuropathol. Exp. Neurol. 53 (1994) 438–447.
Masters, K. Beyreuther, The amyloid precursor protein of Alzheim- [37] D.J. Selkoe, Normal and abnormal biology of the beta-amyloid er’s disease in the reduction of copper(II) to copper(I), Science 271 precursor protein, Annu. Rev. Neurosci. 17 (1994) 489–517. (1996) 1406–1409. [38] V.L. Smith-Swintosky, L.C. Pettigrew, S.D. Craddock, A.R. Culwell, [24] M. Nedergaard, Transient focal ischemia in hyperglycemic rats is R.E. Rydel, M.P. Mattson, Secreted forms of beta-amyloid precursor associated with increased cerebral infarction, Brain Res. 408 (1987) protein protect against ischemic brain injury, J. Neurochem. 63
79–85. (1994) 781–784.
[25] N. Nukina, I. Kanazawa, T. Mannen, Y. Uchida, Accumulation of [39] D.T. Stephenson, K. Rash, J.A. Clemens, Amyloid precursor protein amyloid precursor protein and beta-protein immunoreactivities in accumulates in regions of neurodegeneration following focal cere-axons injured by cerebral infarct, Gerontology 38 (Suppl. 1) (1992) bral ischemia in the rat, Brain Res. 593 (1992) 128–135. 10–14. [40] T. Suenaga, K. Ohnishi, M. Nishimura, S. Nakamura, I. Akiguchi, J. [26] P. Pahlsson, S.H. Shakin-Eshleman, S.L. Spitalnik, N-linked Kimura, Bundles of amyloid precursor protein-immunoreactive glycosylation of beta-amyloid precursor protein, Biochem. Biophys. axons in human cerebrovascular white matter lesions, Acta Neuro-Res. Commun. 189 (1992) 1667–1673. pathol. (Berl.) 87 (1994) 450–455.
[27] G. Paxinos, C. Watson, The Rat Brain in Stereotaxic Coordinates, [41] H. Tomimoto, I. Akiguchi, H. Wakita, S. Nakamura, J. Kimura, Academic Press, Sydney, 1982. Ultrastructural localization of amyloid protein precursor in the [28] R. Pluta, Experimental model of neuropathological changes charac- normal and postischemic gerbil brain, Brain Res. 672 (1995) 187–
teristic for Alzheimer’s disease, Folia Neuropathol. 35 (1997) 94– 195.
98. [42] H. Tomimoto, H. Wakita, I. Akiguchi, S. Nakamura, J. Kimura,
[29] R. Pluta, E. Kida, A.S. Lossinsky, A.A. Golabek, M.J. Mossakow- Temporal profiles of accumulation of amyloid beta /A4 protein ski, H.M. Wisniewski, Complete cerebral ischemia with short-term precursor in the gerbil after graded ischemic stress, J. Cereb. Blood survival in rats induced by cardiac arrest. I. Extracellular accumula- Flow Metab. 14 (1994) 565–573.
tion of Alzheimer’s beta-amyloid protein precursor in the brain, [43] H. Wakita, H. Tomimoto, I. Akiguchi, K. Ohnishi, S. Nakamura, J. Brain Res. 649 (1994) 323–328. Kimura, Regional accumulation of amyloid beta /A4 protein pre-[30] A. Popa-Wagner, E. Schroder, L.C. Walker, C. Kessler, Beta- cursor in the gerbil brain following transient cerebral ischemia,
amyloid precursor protein and ss-amyloid peptide immunoreactivity Neurosci. Lett. 146 (1992) 135–138.
in the rat brain after middle cerebral artery occlusion: effect of age, [44] A.R. White, G. Multhaup, F. Maher, S. Bellingham, J. Camakaris, Stroke 29 (1998) 2196–2202. H. Zheng, A.I. Bush, K. Beyreuther, C.L. Masters, R. Cappai, The [31] W.A. Pulsinelli, S. Waldman, D. Rawlinson, F. Plum, Moderate Alzheimer’s disease amyloid precursor protein modulates copper-hyperglycemia augments ischemic brain damage: a neuropathologic induced toxicity and oxidative stress in primary neuronal cultures, J. study in the rat, Neurology 32 (1982) 1239–1246. Neurosci. 19 (1999) 9170–9179.
[32] J. Ren, G.A. Gintant, R.E. Miller, A.J. Davidoff, High extracellular [45] S.P. Wolff, Z.A. Bascal, J.V. Hunt, ‘Autoxidative glycosylation’: free glucose impairs cardiac E–C coupling in a glycosylation-dependent radicals and glycation theory, Prog. Clin. Biol. Res. 304 (1989) manner, Am. J. Physiol. 273 (1997) H2876–H2883. 259–275.
[33] T.C. Saido, M. Yokota, K. Maruyama, W. Yamao-Harigaya, E. Tani, [46] P.S. Yam, J. Patterson, D.I. Graham, T. Takasago, D. Dewar, J. Y. Ihara, S. Kawashima, Spatial resolution of the primary beta- McCulloch, Topographical and quantitative assessment of white amyloidogenic process induced in postischemic hippocampus, J. matter injury following a focal ischaemic lesion in the rat brain, Biol. Chem. 269 (1994) 15253–15257. Brain Res. Protoc. 2 (1998) 315–322.
[34] T. Saitoh, M. Sundsmo, J.M. Roch, N. Kimura, G. Cole, D. [47] F. Zhang, C. Eckman, S. Younkin, K.K. Hsiao, C. Iadecola, Schubert, T. Oltersdorf, D.B. Schenk, Secreted form of amyloid beta Increased susceptibility to ischemic brain damage in transgenic mice protein precursor is involved in the growth regulation of fibroblasts, overexpressing the amyloid precursor protein, J. Neurosci. 17