REFERAT
THALASSEMIA
Pembimbing:dr. Yahya G. Lubis, Sp.A
Disusun Oleh : Ines Marianne Santoso
030.06.127
KEPANITERAAN KLINIK ILMU KESEHATAN ANAK RSUD KOJA
PERIODE 29 November 2010- 5 Februari 2011 FAKULTAS KEDOKTERAN UNIVERSITAS TRISAKTI
LEMBAR PENGESAHAN
Referat yang berjudul “Thalasemia” telah diterima dan disetujui pada tanggal Januari 2011 oleh pembimbing dr. Yahya G. Lubis, Sp.A sebagai salah satu syarat menyelesaikan
Kepaniteraan Klinik Ilmu Kesehatan Anak - Rumah Sakit Umum Daerah Koja
Jakarta, Januari 2011
dr.Yahya G. Lubis, Sp.A
Puji dan syukur saya panjatkan kepada Tuhan yang Maha Esa atas rahmat dan karuniaNya sehingga saya dapat menyelesaikan karya tulis ini. Karya tulis berjudul “Thalassemia” ini dibuat dengan tujuan sebagai salah satu syarat kelulusan dalam Kepaniteraan Klinik Ilmu Kesehatan Anak di Rumah Sakit Umum Daerah Koja. Dalam pembuatan karya tulis ini, saya mengambil referensi dari literatur dan jaringan internet.
Saya mengucapkan terimakasih yang sebesar-besarnya kepada pembimbing saya, dr.Yahya G. Lubis, Sp.A, yang telah memberikan bimbingannya dalam proses penyelesaian karya tulis ini, juga untuk dukungannya baik dalam bentuk moril maupun dalam mencari referensi yang lebih baik.
Selain itu, saya juga mengucapkan terimakasih kepada teman-teman saya yang berada dalam satu kelompok kepaniteraan yang sama, atas dukungan dan bantuan mereka selama saya menjalani kepaniteraan ini. Pengalaman saya dalam kepaniteraan ini akan selalu menjadi suatu inspirasi yang baik. Saya juga mengucapkan rasa terimakasih yang mendalam kepada kedua orangtua saya atas bantuan, dukungan baik secara moril maupun materil, dan kasihnya.
Semoga karya tulis ini dapat bermanfaat bagi siapa saja yang membacanya.
Penulis,
DAFTAR ISI
Lembar Pengesahan 2 Kata Pengantar 3 Daftar isi 4 BAB I PENDAHULUAN 5 BAB II ISI 1. Definisi 6 2. Epidemio logi 6 3. Fisiologi Hematopoesis 9 4. Patofisiol ogi 16 5. Klasifikas i 21 6. GejalaKlinis (Stadium Thalassemia) 28
7. Diagnosis Banding 28 8. Pemeriks aan Penunjang 30 9. Komplika si 33
10. Terapi 33 11. Skrinning 36 12. Prognosis 37
BAB III KESIMPULAN 38
Daftar Pustaka 39
BAB I
PENDAHULUAN
Thalassemia adalah kelainan bawaan dari sintesis hemoglobin. Presentasi klinisnya bervariasi dari asimtomatik sampai berat hingga mengancam jiwa. Dahulu dinamakan sebagai Mediterannian anemia, diusulkan oleh Whipple, namun kurang tepat karena sebenarnya kondisi ini dapat ditemukan di mana saja di seluruh dunia. Seperti yang akan dijelaskan selanjutnya, beberapa tipe berbeda dari thalassemia lebih endemik pada area geografis tertentu.
Pada tahun 1925, Thomas Cooley, seorang spesialis anak dari Detroit, mendeskripsikan suatu tipe anemia berat pada anak-anak yang berasal dari Italia. Beliau menemukan adanya nukleasi sel darah merah yang masif pada sapuan apus darah tepi, yang mana awalnya beliau pikir sebagai anemia eritroblastik, suatu keadaan yang disebutkan oleh von Jaksh sebelumnya.
Namun tak lama kemudian, Cooley menyadari bahwa eritroblastemia tidak spesifik dan esensial pada temuan ini sehingga istilah anemia eritroblastik tidak dapat dipakai. Meskipun Cooley curiga akan adanya pengaruh genetik dari kelainan ini, namun beliau gagal dalam menginvestigasi orangtua sehat pada anak-anak yang mengidap kelainan ini.
Di Eropa, Riette mendeskripsikan mengenai adanya anemia mikrositik hipokromik ringan yang tak terjelaskan pada anak-anak keturunan Italia pada tahun yang sama saat Cooley melaporan adanya bentuk anemia berat yang akhirnya dinamakan mengikutinya namanya. Sebagi tambahan, Wintrobe di Amerika Serikat melaporkan adanya anemia ringan pada kedua orangtua dari anak yang mengidap anemia Cooley. Anemia ini sangat mirip dengan kelainan yang ditemukan Riette. Baru setelah itu anemia Cooley dinyatakan sebagai bentuk homozigot dari anemia hipokromik mikrositik ringan yang dideskripsikan oleh Riette dan Wintrobe. Bentuk anemia berat ini kemudian dilabelisasi sebagai thalassemia mayor dan bentuk ringannya dinamakan sebagai thalassemia minor. Kata thalassemia berasal dari bahasa Yunani yaitu thalassa yang berarti ‘laut’ (mengarah ke Mediterania), dan emia, yang berarti ‘berhubungan dengan darah’.
BAB II
PEMBAHASAN THALASSEMIA
1. DEFINISI (1)Thalassemia adalah sekelompok heterogen anemia hipokromik herediter dengan berbagai derajat keparahan. Defek genetik yang mendasari meliputi delesi total atau parsial gen globin dan substitusi, delesi, atau insersi nukleotida. Akibat dari berbagai perubahan ini adalah penurunan atau tidak adanya mRNA bagi satu atau lebih rantai globin atau pembentukan mRNA yang cacat secara fungsional. Akibatnya adalah penurunan dan supresi total sintesis rantai polipeptida Hb. Kira-kira 100 mutasi yang berbeda telah ditemukan mengakibatkan fenotip thalassemia; banyak
di antara mutasi ini adalah unik untuk daerah geografi setempat. Pada umumnya, rantai globin yang disintesis dalam eritrosit thalassemia secara struktural adalah normal. Pada bentuk thalassemia-α yang berat, terbentuk hemoglobin hemotetramer abnormal (β4 atau γ4) tetapi komponen polipeptida globin mempunyai struktur normal. Sebaliknya, sejumlah Hb abnormal juga menyebabkan perubahan hemotologi mirip thalassemia.
2. EPIDEMIOLOGI
Di seluruh dunia, 15 juta orang memiliki presentasi klinis dari thalassemia. Fakta ini mendukung thalassemia sebagai salah satu penyakit turunan yang terbanyak; menyerang hampir semua golongan etnik dan terdapat pada hampir seluruh negara di dunia.(2)
Beberapa tipe thalassemia lebih umum terdapat pada area tertentu di dunia. Talasemia α o ditemukan terutama di Asia Tenggara dan kepulauan Mediterania, talasemia α + tersebar di Afrika, Mediterania, Timor Tengah, India dan Asia Tenggara. Angka kariernya mencapai 40-80%.
Thalassemia β memiliki distribusi sama dengan thalassemia α Dengan kekecualian di beberapa negara, frekuensinya rendah di Afrika, tinggi di mediterania dan bervariasi di Timor Tengah, India dan Asia Tenggara. HbE yang merupakan varian thalassemia sangat banyak dijumpai di India, Birma dan beberapa negara Asia Tenggara. Adanya interaksi HbE dan thalassemia β β menyebabkan thalassemia HbE sangat tinggi di wilayah ini.
Yayasan Thalassemia Indonesia menyebutkan bahwa setidaknya 100.000 anak lahir di dunia dengan Thalassemia mayor. Di Indonesia sendiri, tidak kurang dari 1.000 anak kecil menderita penyakit ini. Sedang mereka yang tergolong thalassemia trait jumlahnya mencapai sekitar 200.000 orang.
Di RSCM sampai dengan akhir tahun 2003 terdapat 1060 pasien thalassemia mayor yang berobat jalan di Pusat Thalassemia Departemen Anak FKUI-RSCM yang terdiri dari 52,5 % pasien thalassemia β homozigot, 46,2 % pasien thalassemia HbE, serta thalassemia α 1,3%. Sekitar 70-80 pasien baru, datang tiap tahunnya. (4)
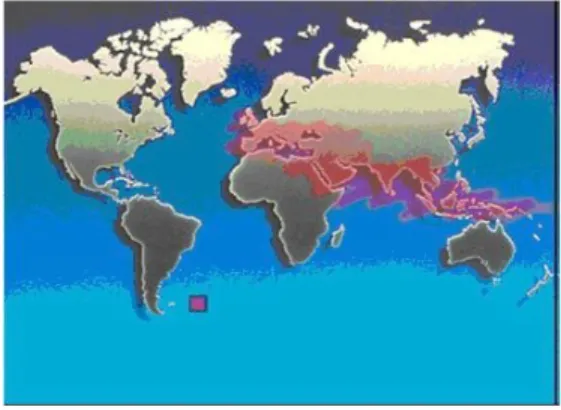
Gambar 1. Daerah Penyebaran Thalassemia/Sabuk Thalassemia.(2)
Mortalitas dan Morbiditas(2)
Thalassemia-α mayor adalah penyakit yang mematikan, dan semua janin yang terkena akan lahir dalam keadaan hydrops fetalis akibat anemia berat. Beberapa laporan pernah mendeskripsikan adanya neonatus dengan thalassemia-α mayor yang bertahan setelah mendapat transfusi intrauterin. Penderita seperti ini membutuhkan perawatan medis yang ekstensif setelahnya, termasuk transfusi darah teratur dan terapi khelasi, sama dengan penderita thalassemia-β mayor. Terdapat juga laporan kasus yang lebih jarang mengenai neonatus dengan thalassemia-α mayor yang lahir tanpa hydrops fetalis yang bertahan tanpa transfusi intrauterin. Pada kasus ini, tingginya level Hb Portland, yang merupakan Hb fungsional embrionik, diperkirakan sebagai penyebab kondisi klinis yang jarang tersebut.
Pada pasien dengan berbagai tipe thalassemia-β, mortalitas dan morbiditas bervariasi sesuai tingkat keparahan dan kualitas perawatan. Thalassemia-β mayor yang berat akan berakibat fatal bila tidak diterapi. Gagal jantung akibat anemia berat atau iron overload adalah penyebab tersering kematian pada penderita. Penyakit hati, infeksi fulminan, atau komplikasi lainnya yang dicetuskan oleh penyakit ini atau terapinya termasuk merupakan penyebab mortalitas dan morbiditas pada bentuk thalassemia yang berat.
Mortalitas dan morbiditas tidak terbatas hanya pada penderita yang tidak diterapi; mereka yang mendapat terapi yang dirancang dengan baik tetap berisiko mengalami bermacam-macam komplikasi. Kerusakan organ akibat iron overload, infeksi berat yang kronis yang dicetuskan transfusi darah, atau komplikasi dari terapi khelasi, seperti katarak, tuli, atau infeksi, merupakan komplikasi yang potensial.
Ras(2)
Meskipun thalassemia ditemukan pada semua ras dan etnik grup, ada beberapa tipe thalassemia yang sering ditemukan pada grup tertentu dibanding dengan yang lain. β thalassemia biasa ditemukan di Eropa Selatan, Timur Tengah, India, dan Africa. α thalassemia biasa ditemukan di Asia Tenggara; meskipun juga ditemukan di bagian dunia yang lain. Mutasi spesifik pada thalassemia sudah dapat discrenning dan didiagnostik kelainannya. α thalassemia trait di Afrika is biasanya bukan dari cis-delesi dari kromosom 16, berbeda dengan di Asia Tenggara, dimana terjadi komplit absence dari α gene pada salah satu chromosome. Pada kedua orang tua yang memiliki cis-delesi, bayinya bias saja mengalami hydrps fetalis. Karena alasan ini, hydops fetalis tidak beresiko tinggi oada rang Afrika tetapi beresiko tinggi pada Asia Tenggara.
Sex (2)
Baik pria maupun wanita,keduanya memiliki kemungkinan yang sama
Usia(2)
Meskipun thalassemia merupakan penyakit turunan (genetik), usia saat timbulnya gejala bervariasi secara signifikan. Dalam talasemia, kelainan klinis pada pasien dengan kasus-kasus yang parah dan temuan hematologik pada pembawa (carrier) tampak jelas pada saat lahir. Ditemukannya hipokromia dan mikrositosis yang tidak jelas penyebabnya pada neonatus, digambarkan di bawah ini, sangat mendukung diagnosis.
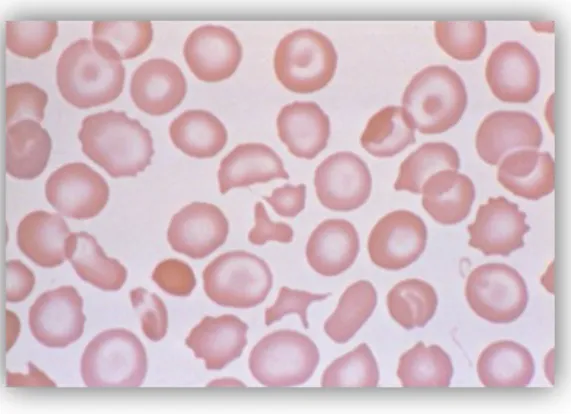
Gambar 2. Sapuan apus darah tepi Penyakit Hb H pada neonatus
Namun, pada thalassemia-β berat, gejala mungkin tidak jelas sampai paruh kedua tahun pertama kehidupan; sampai waktu itu, produksi rantai globin γ dan penggabungannya ke Hb Fetal dapat menutupi gejala untuk sementara.
Bentuk thalassemia ringan sering ditemukan secara kebetulan pada berbagai usia. Banyak pasien dengan kondisi thalassemia-β homozigot yang jelas (yaitu, hipokromasia, mikrositosis, elektroforesis negatif untuk Hb A, bukti bahwa kedua orang tua terpengaruh) mungkin tidak menunjukkan gejala atau anemia yang signifikan selama beberapa tahun. Hampir semua pasien dengan kondisi tersebut dikategorikan sebagai thalassemia-β intermedia. Situasi ini biasanya terjadi jika pasien mengalami mutasi yang lebih ringan, yaitu gabungan heterozygote for B+ dan B -0 thalssemia, atau gabungan dengan heterozygote yang lain.
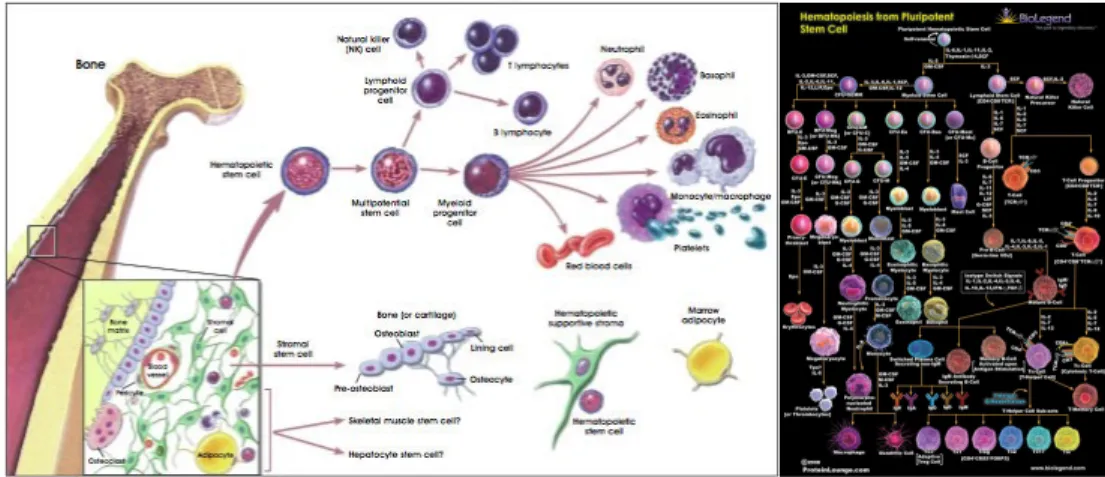
3. FISIOLOGI HEMATOPOESIS
Maximow (1924) mengemukakan suatu dalil bahwa sel darah berasal dari satu sel induk. Hal ini kemudian dikembangkan oleh Downey (1938) yang membuat hipotesa dengan konsep hirarki dari sel pluripoten dan selanjutnya Till dan Mc Culloch (1961) menyimpulkan bahwa satu sel induk merupakan koloni yang memperlihatkan diferensiasi multilineage atau pluripoten menjadi eritroid, mieloid serta megakariosit. Dari penelitian-penelitian tersebut ditetapkan bahwa sel stem ada pada hematopoisis. Sistem hematopoitik mempunyai karakteristik berupa pergantian sel yang konstan untuk mempertahankan populasi leukosit, trombosit dan eritrosit.(3)
Sistem hematopoitik dibagi menjadi 3, yaitu:
2. Colony forming unit (CFU) sebagai pelopor yang selanjutnya berkembang dan berdiferensiasi dalam memproduksi sel.
3. Faktor regulator yang mengatur agar Sistem berlangsung beraturan.
Sel Stem merupakan satu sel induk (klonal) yang mempunyai kemampuan berdiferensiasi menjadi beberapa turunan, membelah diri dan memperbaharui populasi sel stem sendiri di bawah pengaruh faktor pertumbuhan hematopoitik.Hematopoitik membutuhkan perangsang untuk pertumbuhan koloni granulosit dan makrofag yang disebut "Colony Stimulating
Fac-tor" (CSF) yang merupakan glikoprotein.
Dalam proses selanjutnya diketahui regulasi hematopoisis sangat kompleks dan factor pertumbuhan yang berfungsi tumpang tindih serta banyak tempat untuk memproduksi factor-faktor tersebut, termasuk organ hematopoitik. (3)
Dikenal sejumlah sitokin yang mempunyai peranan dalam meningkatkan aktifitas hematopoitik (Tabel 1.1 Faktor pertumbuhan hematopoiesis serta karakterisitiknya)
Faktor Sel Stimulasi Sumber Produksi
Lokasi Kromosom
CS1 (M-CSF) Monosit Sel endotel,
monosit, fibroblast
5q33-1
GM-CSF Granulosit, megakariosit eritrosit,sel stem, blas leukemik
Sel T, sel endotel,
fibroblast
5q23-31
G-CSF Granulosit, makrofag, sel endotelial, fibroblas, blas leukemia
Sel endotel, plasenta,
monosit
17q11-22
IL-3 Granulosit, sel eritroid progenitor, multipoten, blas leukemia
Sel T 5q23-31
IL-5 Sel B, CFU-Eo Sel T 5q31
IL-6 Sel B, CFU-GEMM, CFU
GM, BFU-E, makrofag, sel sel saraf, hepatosit
Fibroblas, leukosit, sel epitel
7p15
IL-7 Sel B Leukosit 8q-12-13
IL-8 Sel T, neutrofil Leukosit 4
IL-9 BFU-E, CFU-GEMM Limfosit 5q31
IL=11 Sel B, T, CFU-GEMM, Makrofag
Makrofag 7q11-22
Eritropoietin CFU-E, BFU-E Ginjal, hepar 7q11-22 c-kit figand
"stem cell factor"
Progenitor primitif NI NI
GM-CSF = granulocyte macrophage colony stimulating factor, G-CSF= granulocyte colony stimulating factor, IL=interleukin, BFU-E=burst forming unit erithrocyte, CFU -E= colony forming unit erythrocyte, CFU-GEMM= colony forming unit granulocyte, erythrocyte, macrophage monocyte, CFU-GM= colony forming unit netrophil-macrophage(3)
Pembentukan dan asal darah (3)
Perkembangan sistem vaskuler dan hematopoisis dimulai pada awal kehidupan embrio dan berlangsung secara paralel / bersamaan sampai masa dewasa mempunyai hubungan dengan lokasi anatomi yang menyokong hematopoisis tersebut.
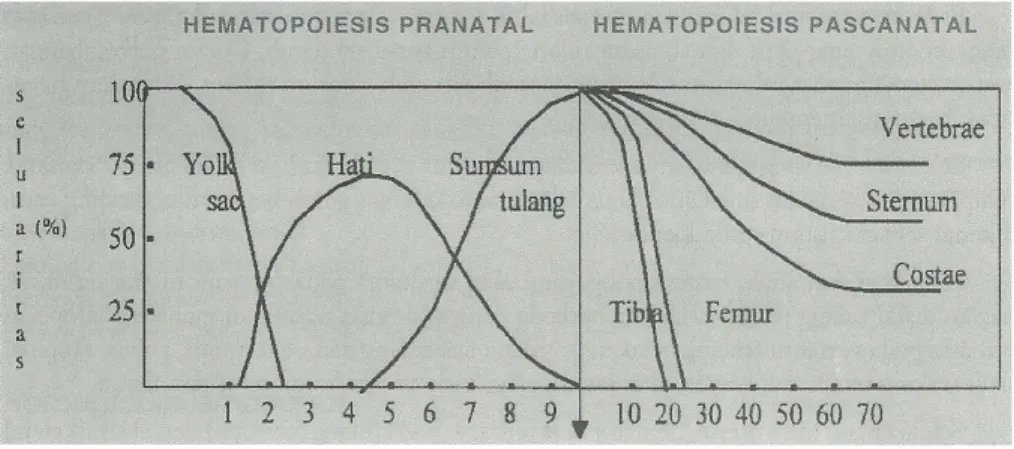
Secara garis besar perkembangan hematopoisis dibagi dalam 3 periode: 1. Hematopoisis yolk sac (mesoblastik atau primitif )
3. Hematopoisis medular
Gambar 3. Hematopoiesis prenatal dan postnatal (dikutip dari Hasan R,1985)
Hematopoisis Yolk Sac (mesoblastik atau primitif)
Sel darah dibuat dari jaringan mesenkim 2-3 minggu selelah fertilisasi. Mula-mula terbentuk dalam blood island yang merupakan pelopor dari sistem vaskuler dan hematopoisis. Selanjutnya eritrosit dan megakariosit dapat diidentifikasi dalam yolk sac pada masa gestasi 16 hari.
Sel induk primitif hematopoisis berasal dari mesoderm mempunyai respons terhadap faktor pertumbuhan antara lain eritropoetin, IL-3, IL-6 dan faktor sel stem. Sel induk hematopoisis mulai berkelompok dalam hati janin pada masa gestasi 5-6 minggu dan pada masa gestasi 8 minggu blood island mengalami regresi. (3)
Hematopoisis hati (Definitif)
Hematopoisis hati berasal dari sel stem pluripoten yang berpindah dari yolk sac. Perubahan empat hematopoisis dari yolk sac ke hati dan kemudian sumsum tulang mempunyai hubungan dengan regulasi perkembangan oleh lingkungan mikro, produksi sitokin dan komponen merangsang adhesi dari matrik ekstraseluler dan ekspresi pada reseptor.
Pada masa gestasi 9 minggu, hematopoisis sudah terbentuk dalam hati. Hematopoisis dalam hati yang terutama adalah eritropoisis, walaupun masih ditemukan sirkulasi granulosit dantrombosit. Hematopoisis hati mencapai puncaknya pada masa gestasi 4-5 bulan kemudian mengalami regresi perlahan-lahan. Pada masa pertengahan kehamilan, tampak pelopor hematopoetik terdapat di limpa, thymus, kelenjar limfe dan ginjal. (3)
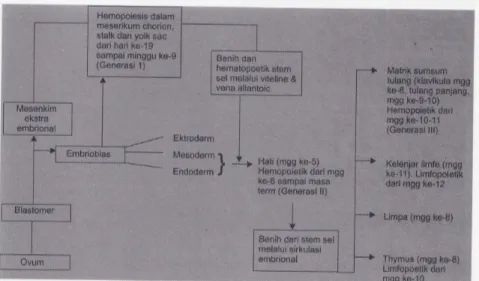
Gambar 4. Perkembangan embrional dan fetal serta ontogeni hematopoesis ( dikutip dari Hasan, 1985)
Hematopoisis medular
Merupakan periode terakhir pembentukan sistem hematopoisis dan dimulai sejak masa gestasi ulan. Ruang medular terbentuk dalam tulang rawan dan tulang panjang dengan proses reabsorpsi.
Pada masa gestasi 32 minggu sampai lahir, semua rongga sumsum tulang diisi jaringan hematopoitik yang aktif dan sumsum tulang penuh berisi sel darah. Dalam perkembangan selanjutnya fungsi pembuatan sel darah diambil alih oleh sumsum tulang, sedangkan hepar tidak berfungsi membuat sel darah lagi. (3)
Sel mesenkim yang mempunyai kemampuan untuk membentuk sel darah menjadi kurang, tetapi tetap ada dalam sumsum tulang, hati, limpa, kelenjar getah bening dan dinding sus, dikenal sebagai sistem retikuloendotelial.
Pada bayi dan anak, hematopoisis yang aktif terutama pada sumsum tulang termasuk bagian distal tulang panjang. Hal ini berbeda dengan dewasa normal di mana hematopoisis terbatas pada vertebra (tulang belakang), tulang iga, tulang dada (sternum), pelvis, skapula, skull (tulang tengkorak kepala) dan jarang yang berlokasi pada humerus dan femur.
Selama masa intra uterin, hematopoisis terdapat pada tulang (skeletal) dan ekstraskeletal dan pada waktu lahir hematopoisis terutama pada skeletal. Secara umum hematopoisis ekstra medular terutama pada organ perut, terjadi akibat penyakit yang menyebabkan gangguan produksi satu atau lebih tipe sel darah, seperti eritroblastosis
fetalis, anemia pernisiosa, talasemia, nickel cell anemia, sferositosis herediter dan variasi leukemia.
Perpindahan lokasi anatomi hematopoisis disertai perpindahan populasi sel sampai ini belum dapat diketahui mekanismenya. (3)
Gambar 5. Pembentukan sel darah
Hemoglobin(4)
Merupakan kompleks protein yang terdiri dari heme yang mengandung besi dan globin dengan interaksi dianatar heme dan globin menyebabkan hemoglobin (Hb) merupakan perangkat yang ireversibel untuk mengangkut oksigen. Sesuai dengan rangkaian hematopoisis yang dimulai dari yolk sac, limpa, hati dan sumsum tulang diikuti juga dengan variasi sintesis hemoglobin. Sejak masa embrio, janin, anak dan dewasa sel darah merah mempunyai 6 hemoglobin antara lain:
Hemoglobin embrional : Gower-1, Gower-2, Portland Hemoglobin fetal : Hb-F
Hemoglobin dewasa : Hb-A1 dan Hb-A2
Hemoglobin embrional(4)
Selama masa gestasi 2 minggu pertama, eritroblas priomitif dalam yolk sac membentuk rantai globin-epsilon (ε ) dan zeta (Z) yang akan membentuk hemoglobin primitive Gower-1 (Z2ε 2). Selanjutnya mulai sintesis rantai α mengganti rantai zeta; rantai γ mengganti rantai ε di yolk sac, yang akan membentuk Hb-Portland (Z2γ2) dan Gower-2 (α2ε 2)
dan Gower-2 yaitu kira-kira 75% dan merupakan hemoglobin yang disintesis di yolk sac, tetapi akan menghilang pada masa gestasi 3 bulan.
Hemoglobin fetal(4)
Migrasi pluripoten stem cell dari yolk sac ke hati, diikuti dengan sintesis hemoglobin fetal dan awal sintesis rantai β. Setelah masa gestasi 8 minggu Hb-F paling dominan dan setelah janin berusai 6 bulan merupakan 90% dari keseluruhan hemoglobin, kemudian berkurang bertahap dan pada saat lahir ditemukan kira-kira 70% Hb-F. sintesis Hb-F menuurun secara cepat setelah bayi lahir dan setelah usia 6-12 bulan hanya sedikit ditemukan.
Hemoglobin dewasa(4)
Pada masa embrio telah dapat dideteksi HbA (α2β2) karena telah terjadi perubahan sintesis rantai γ menjadi β dan selanjutnya globin β meningkat pada ,masa gestasi 6 bulan ditemukan 5-10% HbA, pada waktu lahir mencapai 30% dan pada usia 6-12 bulan sudah memperlihatkan gambaran hemoglobin dewasa.
Hemoglobin dewasa minor (HbA2) ditemukan kira-kira 1% pada saat lahir dan pada usia 12 bulan mencapai 2-3,4%, dengan rasio normal antara HbA dan HbA2 adalah 30:1.Perubahan hemoglobin janin ke dewasa merupakan proses biologi berupa diferensiasi sel induk eritroid, sel stem pluripoten, gen dan reseptor yang mempengaruhi eritroid dan dikontrol oleh factor humoral.
Gambar 6.
Sintesis rantai globin primitive dan definitive selama periode embrional, fetal dan pascanatal
dalam hubungannya dengan perubahan tempat eritropoisis.
4. PATOFISIOLOGI
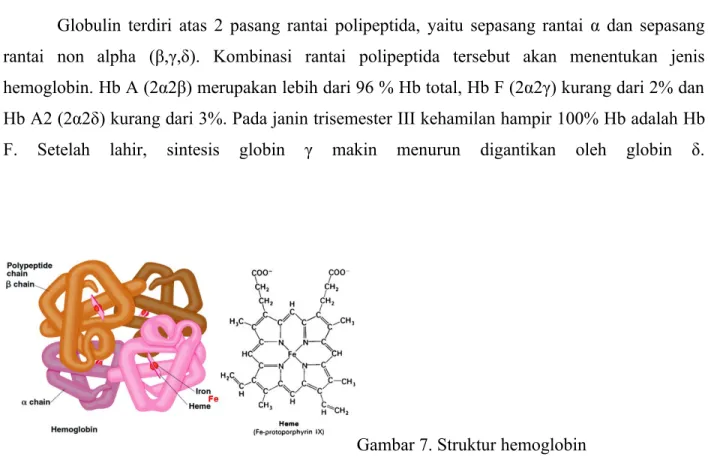
Hemoglobin (Hb) tersusun atas heme yang merupakan cincin porfirin dalam ikatan dengan Fe dan globulin yang merupakan protein pendukung. Satu molekul hemoglobin
mengandung 4 sub-unit. Masing-masing sub-unit tersusun atas satu molekul globin dan satu molekul heme.
Globulin terdiri atas 2 pasang rantai polipeptida, yaitu sepasang rantai α dan sepasang rantai non alpha (β,γ,δ). Kombinasi rantai polipeptida tersebut akan menentukan jenis hemoglobin. Hb A (2α2β) merupakan lebih dari 96 % Hb total, Hb F (2α2γ) kurang dari 2% dan Hb A2 (2α2δ) kurang dari 3%. Pada janin trisemester III kehamilan hampir 100% Hb adalah Hb F. Setelah lahir, sintesis globin γ makin menurun digantikan oleh globin δ.
Gambar 7. Struktur hemoglobin
Rantai polipeptida α tersusun atas 141 asam amino, sedangkan rantai non α tersusun atas 146 asam amino. Sintesis rantai α disandi oleh gen α1 dan gen α2 di kromosom 16, sedangkan gen yang mensintesis rantai β, rantai γ dan rantai δ terletak di kromosom 11. Pada orang normal sintesis rantai α sama dengan rantai non alpha. Thalassemia akan terjadi bila sintesis salah satu rantai polipeptida menurun.
Struktur kimia hemoglobin memungkinkan molekul hemoglobin memiliki kemampuan untuk mengikat oksigen secara reversible. Zat besi dalam molekul heme secara langsung
berfungsi sebagai pengikat oksigen. Hemoglobin memiliki struktur kuartener empat rantai polipeptida, masing-masing dengan satu tempat pegikatan oksigen. Sehingga satu molekul hemoglobin dapat mengikat 4 molekul oksigen. Hemoglobin yang merupakan suatu protein, disintesis berdasarkan informasi genetik. Masing-masing polipeptida penyusun Hb berbeda dalam urutan asam aminonya. Dengan demikian ada beberapa lokus gen terpisah dalam kromosom yang mengatur sintesis rantai polipeptida dari hemoglobin. (2)
Lokus α β γ δ
Genotip
α/α β/β γ/γ δ/δ
Polipetida yang terbentuk α β γ δ
Hb yang terbentuk α2β2 α2γ2 α2δ2
Untuk pembentukan α dan γ sebenarnya terdapat 2 lokus gen untuk masing-masing, sedangkan β dan δ hanya memilki satu lokus gen. Lokus gen untuk α terletak pada kromosom 16 sedangkan lainnya (β,γ,δ) terletak pada kromosom 11.
Sintesis rantai γ bersama dengan sintesi rantai menonjol selama masa kehidupan janin. Rantai α akan terus disintesis sampai usia dewasa sedangkan rantai γ mulai menurun pada trisemester akhir dan dengan cepat menurun setelah kelahiran.
Talasemia merupakan salah satu bentuk kelainan genetik hemoglobin yang ditandai dengan kurangnya atau tidak adanya sintesis satu rantai globin atau lebih, sehingga terjadi ketidak seimbangan jumlah rantai globin yang terbentuk.
Secara genetik, gangguan pembentukan protein globin dapat disebabkan karena kerusakan gen yang terdapat pada kromosom 11 atau 16 yang ditempati lokus gen globin. Sebagian besar kelainan hemoglobin dan jenis thalassemia merupakan hasil kelaianan mutasi pada gamet yang terjadi pada replikasi DNA. Pada replikasi DNA dapat terjadi pergantian urutan asam basa dalam DNA, dan perubahan kode genetic akan diteruskan pada penurunan genetic berikutnya. Mutasi ini dapat memperpendek rantai asam amino maupun memperpanjangnya. Kelainan mutasi dapat pula terjadi pada keselahan berpasangan kromosom pada proses meiosis yang mengakibatkan perubahan susunan material genetic. Bila terjadi crossing over pada kesalahan berpasangan itu, sebagai hasil akhir peristiwa tadi akan terjjadi apa yang disebut duplikasi,delesi, translokasi dan iversi. Kerusakan pada salah satu kromosom homolog menimbulkan terjadinya keadaan heterozigot, sedangkan kerusakan pada kedua kromosom homolog menimbulkan keadaan homozigot.
Pada thalassemia homozigot sintesis rantai menurun atau tidak ada sintesis sama sekali. Ketidakseimbangan sintesis rantai alpha atau rantai non alpha, khususnya kekurangan sintesis rantai β akan menyebabkan kurangnya pembentukan Hb.
Ketidakseimbangan dalam rantai protein globin alfa dan beta, yang diperlukan dalam pembentukan hemoglobin, disebabkan oleh sebuah gen cacat yang diturunkan. Untuk menderita penyakit ini, seseorang harus memiliki 2 gen dari kedua orang tuanya. Jika hanya 1 gen yang diturunkan, maka orang tersebut hanya menjadi pembawa tetapi tidak menunjukkan gejala-gejala dari penyakit ini. (2)
Secara biokimia kelainan yang paling mendasar adalah menurunnya biosintesis dari unit β globin pada Hb A. pada thalasemia β heterozigot, sintesis β globin kurang lebih separuh dari nilai normalnya. Pada thalasemia β homozigot, sintesis β globin dapat mencapai nol.
Karena adanya defisiensi yang berat pada rantai β, sintesis Hb A total menurun dengan sangat jelas atau bahkan tidak ada, sehingga pasien dengan thalasemia β homozigot mengalami anemia berat. Sebagai respon kompensasi, maka sintesis rantai γ menjadi teraktifasi sehingga
hemoglobin pasien mengandung proporsi Hb F yang meningkat. Namun sintesis rantai γ ini tidak efektif dan secara kuantitas tidak mencukupi. (7)
Pada thalasemia β homozigot, sintesis rantai α tidak mengalami perubahan. Ketidak-seimbangan sintesis dari rantai polipeptida ini mengakibatkan kelebihan adanya rantai α bebas di dalam sel darah merah yang berinti dan retikulosit. Rantai α bebas ini mudah teroksidasi. Mereka dapat beragregasi menjadi suatu inklusi protein (haeinz bodys), menyebabkan kerusakan membran pada sel darah merah dan destruksi dari sel darah merah imatur dalam sumsum tulang sehingga jumlah sel darah merah matur yang diproduksi menjadi berkurang. Sel darah merah yang beredar kecil, terdistorsi, dipenuhi oleh inklusi α globin, dan mengandung komplemen hemoglobin yang menurun. Hal yang telah disebutkan diatas adalah gambaran dari Anemia Cooley: hipokromik, mikrosisitk dan poikilositik.
Sel darah merah yang sudah rusak tersebut akan dihancurkan oleh limpa, hepar, dan sumsum tulang, menggambarkan komponen hemolitik dari penyakit ini. Sel darah merah yang mengandung jumlah Hb F yang lebih tinggi mempunyai umur yang lebih panjang.
Anemia yang berat terjadi akibat adanya penurunan oksigen carrying capacity dari setiap eritrosit dan tendensi dari sel darah merah matur (yang jumlahnya sedikit) mengalami hemolisa secara prematur.
Eritropoetin meningkat sebagai respon adanya anemia, sehingga sumsum-sumsum tulang dipacu untuk memproduksi eritroid prekusor yang lebih banyak. Namun mekanisme kompensasi ini tidak efektif karena adanya kematian yang prematur dari eritroblas. Hasilnya adalah suatu ekspansi sumsum tulang yang masif yang memproduksi sel darah merah baru.
Sumsum tulang mengalami ekspansi secara masif, menginvasi bagian kortikal dari tulang, menghabiskan sumber kalori yang sangat besar pada umur-umur yang kritis pada pertumbuhan dan perkembangan, mengalihkan sumber-sumber biokimia yang vital dari tempat-tempat yang membutuhkannya dan menempatkan suatu stress yang sangat besar pada jantung. Secara klinis terlihat sebagai kegalan dari pertumbuhan dan perkembangan, kegagalan jantung high output, kerentanan terhadap infeksi, deformitas dari tulang, fraktur patologis, dan kematian di usia muda tanpa adanya terapi transfusi. (8)
Dengan pemberian transfusi darah, eritropoesis yang inefektif dapat diperbaiki, dan terjadi peningkatan jumlah hormon hepcidin; sehingga penyerapan besi akan berkurang dan makrofag akan mempertahankan kadar besi.
Pada pasien dengan iron overload (misalnya hemokromatosis), absorpsi besi menurun akibat meningkatnya jumlah hepsidin. Namun, hal ini tidak terjadi pada penderita thalassemia-β berat karena diduga faktor plasma menggantikan mekanisme tersebut dan mencegah terjadinya produksi hepsidin sehingga absorpsi besi terus berlangsung meskipun penderita dalam keadaan iron overload.
Efek hepsidin terhadap siklus besi dilakukan melalui kerja hormon lain bernama ferroportin, yang mentransportasikan besi dari enterosit dan makrofag menuju plasma dan menghantarkan besi dari plasenta menuju fetus. Ferroportin diregulasi oleh jumlah penyimpanan besi dan jumlah hepsidin. Hubungan ini juga menjelaskan mengapa penderita dengan thalassemia-β yang memiliki jumlah besi yang sama memiliki jumlah ferritin yang berbeda sesuai dengan apakah mereka mendapat transfusi darah teratur atau tidak. Sebagai contoh, penderita thalassemia-β intermedia yang tidak mendapatkan transfusi darah memiliki jumlah ferritin yang lebih rendah dibandngkan dengan penderita yang mendapatkan transfusi darah secara teratur, meskipun keduanya memiliki jumlah besi yang sama.
Kebanyakan besi non-heme pada individu yang sehat berikatan kuat dengan protein pembawanya, transferrin. Pada keadaan iron overload, seperti pada thalassemia berat, transferrin tersaturasi, dan besi bebas ditemukan di plasma. Besi ini cukup berbahaya karena memiliki material untuk memproduksi hidroksil radikal dan akhirnya akan terakumulasi pada organ-organ, seperti jantung, kelenjar endokrin, dan hati, mengakibatkan terjadinya kerusakan pada organ-organ tersebut (organ damage). (2)
5. KLASIFIKASI
Talasemia adalah grup kelainan sintesis hemoglobin yang heterogen akibat pengurangan produksi satu atau lebih rantai globin. Hal ini menyebabkan ketidakseimbangan produksi rantai globin.
Sebagaimana telah disebutkan di atas, secara garis besar terdapat dua tipe utama thalassemia yaitu α thalassemia dan β thalassemia. Selain itu juga terdapat tipe thalassemia lain seperti thalassemia intermediate.
Abnormalitas genetic Sindroma klinik Thalassemia α
Penghapusan 4 gen- hydrops fetalis Penghapusan 3 gen- penyakit Hb H Penghapusan 2 gen ( trait thalasemia α° ) Penghapusan 1 gen ( trait thalasemia α+ )
Kematian in utero Anemia hemolitik
Sediaan darah mikrositik hipokrom tetapi biasanya tanpa anemia
Thalassemia β
Homozigot – thalassemia mayor Heterzigot- trait thalassemia
Anemia berat perlu transfusi darah
Sediaan darah mikrositik hipokrom tetapi biasanya dengan atau tanpa anemia
Thalassemia intermediate
Sindroma klinik yang disebabkan oleh sejenis lesi genetik
Anemia hipokrom mikrositik, hepato- splenomegali, kelebihan beban besi.
Talasemia diturunkan berdasarkan hukum Mendel, resesif atau ko-dominan. Heterozigot biasanya tanpa gejala homozigot atau gabungan heterozigot gejalanya lebih berat dari talasemia α atau β .(2)
Thalassemia-α(7)
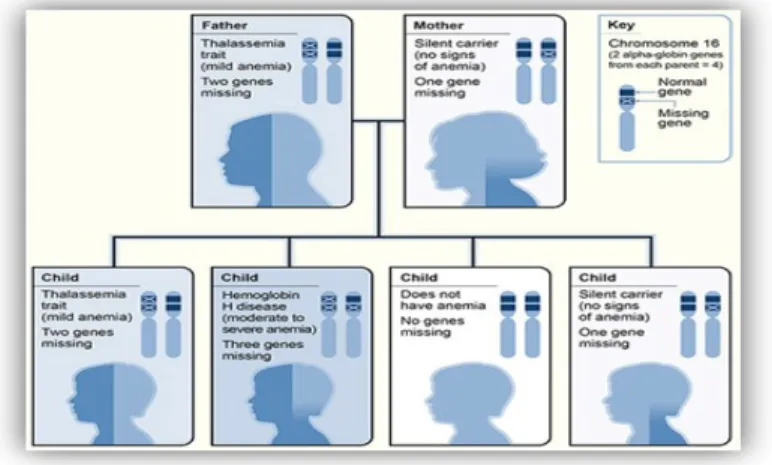
Anemia mikrositik yang disebabkan oleh defisiensi sintesis globin-α banyak ditemukan di Afrika, negara di daerah Mediterania, dan sebagian besar Asia. Delesi gen globin-α menyebabkan sebagian besar kelainan ini. Terdapat empat gen globin-α pada individu normal, dan empat bentuk thalassemia-α yang berbeda telah diketahui sesuai dengan delesi satu, dua, tiga, dan semua empat gen ini.
Tabel 1. Thalassemia-α
Genotip Jumlah gen α Presentasi Klinis Hemoglobin Elektroforesis Saat Lahir > 6 bulan
αα/αα 4 Normal N N
-α/αα 3 Silent carrier 0-3 % Hb Barts N
--/αα atau –α/-α
2 Trait thal-α 2-10% Hb Barts N
--/-α 1 Penyakit Hb H 15-30% Hb Bart Hb H
--/-- 0 Hydrops fetalis >75% Hb Bart
-Ket : N = hasil normal, Hb = hemoglobin, Hb Bart’s = γ4, HbH = β4
a. Silent carrier thalassemia-α
- Merupakan tipe thalassemia subklinik yang paling umum, biasanya ditemukan secara kebetulan diantara populasi, seringnya pada etnik Afro-Amerika. Seperti telah dijelaskan sebelumnya, terdapat 2 gen α yang terletak pada kromosom 16.
- Pada tipe silent carrier, salah satu gen α pada kromosom 16 menghilang, menyisakan hanya 3 dari 4 gen tersebut. Penderita sehat secara hematologis, hanya ditemukan adanya jumlah eritrosit (sel darah merah) yang rendah dalam beberapa pemeriksaan.
- Pada tipe ini, diagnosis tidak dapat dipastikan dengan pemeriksaan elektroforesis Hb, sehingga harus dilakukan tes lain yang lebih canggih. Bisa juga dicari akan adanya kelainan hematologi pada anggota keluarga (misalnya orangtua) untuk mendukung diagnosis. Pemeriksaan darah lengkap pada salah satu orangtua yang menunjukkan adanya hipokromia dan mikrositosis tanpa penyebab yang jelas merupakan bukti yang cukup kuat menuju diagnosis thalasemia. (7)
b. Trait thalassemia-α
- Trait ini dikarakterisasi dengan anemia ringan dan jumlah sel darah merah yang rendah. Kondisi ini disebabkan oleh hilangnya 2 gen α pada satu kromosom 16 atau satu gen α pada masing-masing kromosom. Kelainan ini sering ditemukan di Asia Tenggara, subbenua India, dan Timur Tengah.
- Pada bayi baru lahir yang terkena, sejumlah kecil Hb Barts (γ4) dapat ditemukan pada elektroforesis Hb. Lewat umur satu bulan, Hb Barts tidak terlihat lagi, dan kadar Hb A2 dan HbF secara khas normal. (7)
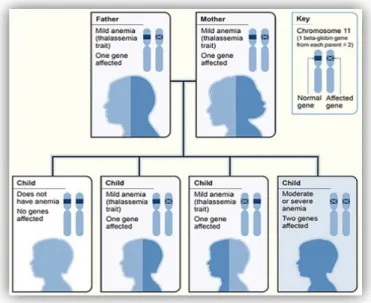
Gambar 7. Thalassemia alpha menurut hukum Mendel (6)
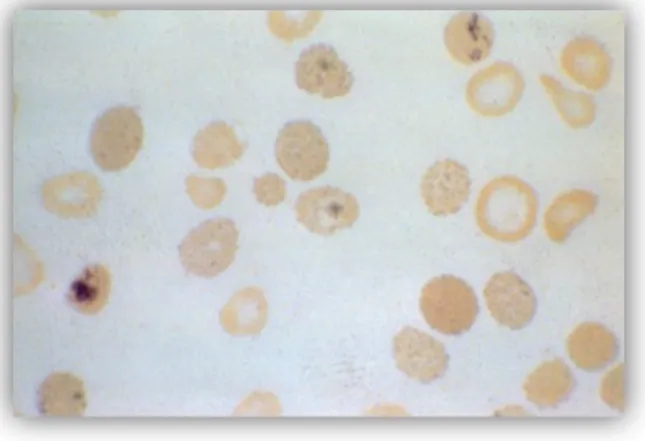
c. Penyakit Hb H
Kelainan disebabkan oleh hilangnya 3 gen globin α, merepresentasikan thalassemia-α intermedia, dengan anemia sedang sampai berat, splenomegali, ikterus, dan jumlah sel darah merah yang abnormal. Pada sediaan apus darah tepi yang diwarnai dengan pewarnaan supravital akan tampak sel-sel darah merah yang diinklusi oleh rantai tetramer
β (Hb H) yang tidak stabil dan terpresipitasi di dalam eritrosit, sehingga menampilkan gambaran golf ball. Badan inklusi ini dinamakan sebagai Heinz bodies. (7)
Gambar 8. Pewarnaan supravital pada sapuan apus darah tepi Penyakit Hb H yang menunjukkan Heinz-Bodies
d. Thalassemia-α mayor
- Bentuk thalassemia yang paling berat, disebabkan oleh delesi semua gen globin-α, disertai dengan tidak ada sintesis rantai α sama sekali.
- Karena Hb F, Hb A, dan Hb A2 semuanya mengandung rantai α, maka tidak satupun dari Hb ini terbentuk. Hb Barts (γ4) mendominasi pada bayi yang menderita, dan karena γ4 memiliki afinitas oksigen yang tinggi, maka bayi-bayi itu mengalami hipoksia berat. Eritrositnya juga mengandung sejumlah kecil Hb embrional normal (Hb Portland = ζ2γ2), yang berfungsi sebagai pengangkut oksigen.
- Kebanyakan dari bayi-bayi ini lahir mati, dan kebanyakan dari bayi yang lahir hidup meninggal dalam waktu beberapa jam. Bayi ini sangat hidropik, dengan gagal jantung kongestif dan edema anasarka berat. Yang dapat hidup dengan manajemen neonatus agresif juga nantinya akan sangat bergantung dengan transfusi. (7)
Thalassemia-β (8)
Sama dengan thalassemia-α, dikenal beberapa bentuk klinis dari thalassemia-β; antara lain :
a. Trait thalassemia-β+ heterozigot (Thalassemia minor)
- Penderita mengalami anemia ringan, nilai eritrosit abnormal, dan elektroforesis Hb abnormal dimana didapatkan peningkatan jumlah Hb A2, Hb F, atau keduanya.
- Individu dengan ciri (trait) thalassemia sering didiagnosis salah sebagai anemia defisiensi besi dan mungkin diberi terapi yang tidak tepat dengan preparat besi selama waktu yang panjang. Lebih dari 90% individu dengan trait thalassemia-β mempunyai peningkatan Hb-A2 yang berarti (3,4%-7%). Kira-kira 50% individu ini juga mempunyai sedikit kenaikan HbF, sekitar 2-6%. Pada sekelompok kecil kasus, yang benar-benar khas, dijumpai Hb A2 normal dengan kadar HbF berkisar dari 5% sampai 15%, yang mewakili thalassemia tipe δβ. (8)
Gambar 10. Sapuan darah tepi tampak sel target
b. Thalassemia-β° homozigot (Anemia Cooley, Thalassemia Mayor)
- Bergejala sebagai anemia hemolitik kronis yang progresif selama 6 bulan kedua kehidupan. Transfusi darah yang reguler diperlukan pada penderita ini untuk mencegah kelemahan yang amat sangat dan gagal jantung yang disebabkan oleh anemia. Tanpa transfusi, 80% penderita meninggal pada 5 tahun pertama kehidupan.
- Pada kasus yang tidak diterapi atau pada penderita yang jarang menerima transfusi pada waktu anemia berat, terjadi hipertrofi jaringan eritropoetik disumsum tulang maupun di luar sumsum tulang. Tulang-tulang menjadi tipis dan fraktur patologis mungkin terjadi. Ekspansi masif sumsum tulang di wajah dan tengkorak menghasilkan bentuk wajah yang khas.
Gambar 11. Deformitas tulang pada thalassemia beta mayor (Facies Cooley)
- Pucat, hemosiderosis, dan ikterus sama-sama memberi kesan coklat kekuningan. Limpa dan hati membesar karena hematopoesis ekstrameduler dan hemosiderosis. Pada penderita yang lebih tua, limpa mungkin sedemikian besarnya sehingga menimbulkan ketidaknyamanan mekanis dan hipersplenisme sekunder.
Gambar 12. Splenomegali pada thalassemia
- Pertumbuhan terganggu pada anak yang lebih tua; pubertas terlambat atau tidak terjadi karena kelainan endokrin sekunder. Diabetes mellitus yang disebabkan oleh siderosis pankreas mungkin terjadi. Komplikasi jantung, termasuk aritmia dan gagal jantung kongestif kronis yang disebabkan oleh siderosis miokardium sering merupakan kejadian terminal.
- Kelainan morfologi eritrosit pada penderita thalassemia-β° homozigot yang tidak ditransfusi adalah ekstrem. Disamping hipokromia dan mikrositosis berat, banyak ditemukan poikilosit yang terfragmentasi, aneh (sel bizarre) dan sel target. Sejumlah besar eritrosit yang berinti ada di darah tepi, terutama setelah splenektomi. Inklusi intraeritrositik, yang merupakan presipitasi kelebihan rantai α, juga terlihat pasca splenektomi. Kadar Hb turun secara cepat menjadi < 5 gr/dL kecuali mendapat transfusi. Kadar serum besi tinggi dengan saturasi kapasitas pengikat besi (iron binding capacity). Gambaran biokimiawi yang nyata adalah adanya kadar HbF yang sangat tinggi dalam eritrosit. (8)
6. GEJALA KLINIS (STADIUM THALASSEMIA) (9)
Gejala klinis pada thalassemia hampir semua sama, yang membedakan adalah tingkat keparahannya, dari ringan (asimptomatik) sampai parahnya gejala.. Gejala klinis biasa berupa
tanda-tanda anemia seperti pucat, lemah,letih,lesu, tidak aktif beraktifitas atau jarang bermain dengan teman seusianya, sesak nafas kurang konsentrasi, sering pula disertai dengan kesulitan makan, gagal tumbuh, infeksi berulang dan perubahan tulang. Pada pemeriksaan fisik didapatkan facies Cooley, conjungtiva anemis, bentuk tulang yang abnormal, pembesarah lien dan atau hepar.
Terdapat suatu sistem pembagian stadium thalassemia berdasarkan jumlah kumulatif transfusi darah yang diberikan pada penderita untuk menentukan tingkat gejala yang melibatkan kardiovaskuler dan untuk memutuskan kapan untuk memulai terapi khelasi pada pasien dengan thalassemia-β mayor atau intermedia. Pada sistem ini, pasien dibagi menjadi tiga kelompok, yaitu :
1. Stadium I
Merupakan mereka yang mendapat transfusi kurang dari 100 unit Packed Red Cells (PRC). Penderita biasanya asimtomatik, pada echokardiogram (ECG) hanya ditemukan sedikit penebalan pada dinding ventrikel kiri, dan elektrokardiogram (EKG) dalam 24 jam normal.
2. Stadium II
Merupakan mereka yang mendapat transfusi antara 100-400 unit PRC dan memiliki keluhan lemah-lesu. Pada ECG ditemukan penebalan dan dilatasi pada dinding ventrikel kiri. Dapat ditemukan pulsasi atrial dan ventrikular abnormal pada EKG dalam 24 jam.
3. Stadium III
Gejala berkisar dari palpitasi hingga gagal jantung kongestif, menurunnya fraksi ejeksi pada ECG. Pada EKG dalam 24 jam ditemukan pulsasi prematur dari atrial dan ventrikular.
7. DIAGNOSIS BANDING
Thalassemia sering kali didiagnosis salah sebagai anemia defisiensi Fe, hal ini disebabkan oleh karena kemiripan gejala yang ditimbulkan, dan gambaran eritrosit mikrositik
hipokrom. Namun kedua penyakit ini dapat dibedakan, karena pada anemia defisiensi Fe didapatkan : (10)
- Pucat tanpa organomegali - SI rendah
- IBC meningkat
- Tidak tedapat besi dalam sumsum tulang
- Bereaksi baik dengan pengobatan dengan preparat besi
Gambar 13. Apusan darah tepi defisiensi besi
Anemia sideroblastik dimana didaptkan pula gambaran apusan darah tepi mikrositik hipokrom dan gejala-gejala anemia, yang membedakan dengan thalassemia adalah kadar besi dalam darah tinggi, kadar TIBC (Total Iron Binding Capacity) normal atau meningkat sedangkan pada thalassemia kadar besi dan TIBC normal.
Dapat juga dibandingkan dengan anemia defisiensi G6PD, dimana enzim ini bekerja untuk mencegah kerusakan eritrosit akibat oksidasi. Merupakan salah satu anemia hemolitik juga. Dapat dibedakan dengan thalassemia dengan gambaran apusan darah tepi dimana pada defisiensi G6PD nomositik-normokrom dan pemeriksaan enzim G6PD.
Thalassemia juga didiagnosis banding dengan jenis thalassemia lainnya, yang memberi gambaran klinis yang sama. Namun pada pemeriksaan elektroforesis hemoglobin dapat diketahui jenis thalassemia α atau thalassemia β. Pada thalassemia α dengan HbH ditemukan jaundice dan splenomegali. (9)
8. PEMERIKSAAN PENUNJANG
Pemeriksaan laboratorium yang perlu untuk menegakkan diagnosis thalassemia ialah: 1. Darah (2)
Pemeriksaan darah yang dilakukan pada pasien yang dicurigai menderita thalasemia adalah :
- Darah rutin
Kadar hemoglobin menurun. Dapat ditemukan penurunan jumlah eritrosit, peningkatan jumlah lekosit, ditemukan pula peningkatan dari sel PMN. Bila terjadi hipersplenisme akan terjadi penurunan dari jumlah trombosit.
- Hitung retikulosit
Hitung retikulosit meningkat antara 2-8 %. - Gambaran darah tepi
Anemia pada thalassemia mayor mempunyai sifat mikrositik hipokrom. Pada gambaran sediaan darah tepi akan ditemukan retikulosit, poikilositosis, tear drops sel dan target sel.
Gambar 13. Sapuan darah tepi pada thalassemia - Serum Iron & Total Iron Binding Capacity
Kedua pemeriksaan ini dilakukan untuk menyingkirkan kemungkinan anemia terjadi karena defisiensi besi. Pada anemia defisiensi besi SI akan menurun, sedangkan TIBC akan meningkat.
- Tes Fungsi Hepar
Kadar unconjugated bilirubin akan meningkat sampai 2-4 mg%. bila angka tersebut sudah terlampaui maka harus dipikir adanya kemungkinan hepatitis, obstruksi batu empedu dan cholangitis. Serum SGOT dan SGPT akan meningkat dan menandakan adanya kerusakan hepar. Akibat dari kerusakan ini akan berakibat juga terjadi kelainan dalam faktor pembekuan darah. 2. Elektroforesis Hb (2)
Diagnosis definitif ditegakkan dengan pemeriksaan eleltroforesis hemoglobin. Pemeriksaan ini tidak hanya ditujukan pada penderita thalassemia saja, namun juga pada orang tua, dan saudara sekandung jika ada. Pemeriksaan ini untuk melihat jenis hemoglobin dan kadar HbA2. Petunjuk adanya thalassemia α adalah ditemukannya Hb Barts dan Hb H. Pada thalassemia β kadar Hb F bervariasi antara 10-90%, sedangkan dalam keadaan normal kadarnya tidak melebihi 1%.
3. Pemeriksaan sumsum tulang (2)
Pada sumsum tulang akan tampak suatu proses eritropoesis yang sangat aktif sekali. Ratio rata-rata antara myeloid dan eritroid adalah 0,8. pada keadaan normal biasanya nilai perbandingannya 10 : 3.
Gambar 14. Sapuan sumsum tulang May-Giemsa stain, x1000
4. Pemeriksaan rontgen (5)
Ada hubungan erat antara metabolisme tulang dan eritropoesis. Bila tidak mendapat tranfusi dijumpai osteopeni, resorbsi tulang meningkat, mineralisasi berkurang, dan dapat diperbaiki dengan pemberian tranfusi darah secara berkala. Apabila tranfusi tidak optimal terjadi ekspansi rongga sumsum dan penipisan dari korteknya. Trabekulasi memberi gambaran mozaik pada tulang. Tulang terngkorak memberikan gambaran yang khas, disebut dengan “hair on end” yaitu menyerupai rambut berdiri potongan pendek pada anak besar.
Gambar 15. Gmabar rontgen kepala “Hair on end” dan tulang panjang yang terjadi penipisan korteks.
5. EKG dan echocardiography untuk mengetahui dan memonitor keadaan jantungnya. Kadang ditemukan jantung yang kardiomegali akibat anemianya.
6. HLA typing untuk pasien yang akan di transplantasi sumsum tulang.
7. Pemeriksaan mata, pendengaran, fungsi ginjal dan test darah rutin untuk memonitor efek terapi deferoxamine (DFO) dan shelating agent. (9)
- Splenomegali karena penimbunan besi dan eritrosit abnormal, leukosit dan trombosit.
- Anak dengan β thalassemia mayor dengan transfuse yang tidak adekuat dapat menyebabkan pertumbuhan kurang dan mudah terinfeksi, hepatosplenomegali, penipisan cortex tulang dan mudah fraktur.
- Hemosdierosis akibat pemberian transfuse, sehingga kadar serum besi yang berlebihan. - Kerusakan hepar yang disebabkan oleh besi yang berhubungan dengan komplikasi sekunder
dari transfuse dan infeksi hepatitis C merupakan penyebab tersering hepatitis pada anak dengan thalassemia.
- Congestive heart failure dan cardiac aritmia pada transfusi tanpa chelating agent. - Thrombosis dan septikemia pada splenektomi
- Wanita dengan fetus α- thalassemia meningkatkan komplikasi pada kehamilan karena toksikemia dan peradarahan post partum. (10)
9. TERAPI
Penderita trait thalassemia tidak memerlukan terapi ataupun perawatan lanjut setelah diagnosis awal dibuat. Terapi preparat besi sebaiknya tidak diberikan kecuali memang dipastikan terdapat defisiensi besi dan harus segera dihentikan apabila nilai Hb yang potensial pada penderita tersebut telah tercapai. Diperlukan konseling pada semua penderita dengan kelainan genetik, khususnya mereka yang memiliki anggota keluarga yang berisiko untuk terkena penyakit thalassemia berat.
Penderita thalassemia berat membutuhkan terapi medis, dan regimen transfusi darah merupakan terapi awal untuk memperpanjang masa hidup. Transfusi darah harus dimulai pada usia dini ketika anak mulai mengalami gejala dan setelah periode pengamatan awal untuk menilai apakah anak dapat mempertahankan nilai Hb dalam batas normal tanpa transfusi.
a. Transfusi Darah (4)
- Transfusi darah bertujuan untuk mempertahankan nilai Hb tetap pada level 9-9.5 gr/dL sepanjang waktu.
- Pada pasien yang membutuhkan transfusi darah reguler, maka dibutuhkan suatu studi lengkap untuk keperluan pretransfusi. Pemeriksaan tersebut meliputi fenotip sel darah merah, vaksinasi hepatitis B (bila perlu), dan pemeriksaan hepatitis.
- Darah yang akan ditransfusikan harus rendah leukosit; 10-15 mL/kg PRC dengan kecepatan 5 mL/kg/jam setiap 3-5 minggu biasanya merupakan regimen yang adekuat untuk mempertahankan nilai Hb yang diinginkan.
- Pertimbangkan pemberikan asetaminofen dan difenhidramin sebelum transfusi untuk mencegah demam dan reaksi alergi.
Komplikasi Transfusi Darah (4)
Komplikasi utama dari transfusi adalah yang berkaitan dengan transmisi bahan infeksius ataupun terjadinya iron overload. Penderita thalassemia mayor biasanya lebih mudah untuk terkena infeksi dibanding anak normal, bahkan tanpa diberikan transfusi. Beberapa tahun lalu, 25% pasien yang menerima transfusi terekspose virus hepatitis B. Saat ini, dengan adanya imunisasi, insidens tersebut sudah jauh berkurang. Virus Hepatitis C (HCV) merupakan penyebab utama hepatitis pada remaja usia di atas 15 tahun dengan thalassemia. Infeksi oleh organisme opurtunistik dapat menyebabkan demam dan enteriris pada penderita dengan iron overload, khususnya mereka yang mendapat terapi khelasi dengan Deferoksamin (DFO). Demam yang tidak jelas penyebabnya, sebaiknya diterapi dengan Gentamisin dan Trimetoprim-Sulfametoksazol.
b. Terapi Khelasi (Pengikat Besi) (4)
- Apabila diberikan sebagai kombinasi dengan transfusi, terapi khelasi dapat menunda onset dari kelainan jantung dan, pada beberapa pasien, bahkan dapat mencegah kelainan jantung tersebut.
- Chelating agent yang biasa dipakai adalah DFO yang merupakan kompleks hidroksilamin dengan afinitas tinggi terhadap besi. Rute pemberiannya sangat penting untuk mencapai tujuan terapi, yaitu untuk mencapai keseimbangan besi negatif (lebih banyak diekskresi dibanding yang diserap). Karena DFO tidak diserap di usus, maka rute pemberiannya harus melalui parenteral (intravena, intramuskular, atau subkutan).
- Dosis total yang diberikan adalah 30-40mg/kg/hari diinfuskan selama 8-12 jam saat pasien tidur selama 5 hari/minggu.
c. Transplantasi Sel Stem Hematopoetik (TSSH) (4)
TSSH merupakan satu-satunya yang terapi kuratif untuk thalassemia yang saat ini diketahui. Prognosis yang buruk pasca TSSH berhubungan dengan adanya hepatomegali, fibrosis portal, dan terapi khelasi yang inefektif sebelum transplantasi dilakukan. Prognosis bagi penderita yang memiliki ketiga karakteristik ini adalah 59%, sedangkan pada penderita yang tidak memiliki ketiganya adalah 90%. Meskipun transfusi darah tidak diperlukan setelah transplantasi sukses dilakukan, individu tertentu perlu terus mendapat terapi khelasi untuk menghilangkan zat besi yang berlebihan. Waktu yang optimal untuk memulai pengobatan tersebut adalah setahun setelah TSSH. Prognosis jangka panjang pasca transplantasi , termasuk fertilitas, tidak diketahui. Biaya jangka panjang terapi standar diketahui lebih tinggi daripada biaya transplantasi. Kemungkinan kanker setelah TSSH juga harus dipertimbangkan.
d. Terapi Bedah(4)
Splenektomi merupakan prosedur pembedahan utama yang digunakan pada pasien dengan thalassemia. Limpa diketahui mengandung sejumlah besar besi nontoksik (yaitu, fungsi penyimpanan). Limpa juga meningkatkan perusakan sel darah merah dan distribusi besi. Fakta-fakta ini harus selalu dipertimbangkan sebelum memutuskan melakukan splenektomi.. Limpa berfungsi sebagai penyimpanan untuk besi nontoksik, sehingga melindungi seluruh tubuh dari besi tersebut. Pengangkatan limpa yang terlalu dini dapat membahayakan.
Sebaliknya, splenektomi dibenarkan apabila limpa menjadi hiperaktif, menyebabkan penghancuran sel darah merah yang berlebihan dan dengan demikian meningkatkan kebutuhan transfusi darah, menghasilkan lebih banyak akumulasi besi.
Splenektomi dapat bermanfaat pada pasien yang membutuhkan lebih dari 200-250 mL / kg PRC per tahun untuk mempertahankan tingkat Hb 10 gr / dL karena dapat menurunkan kebutuhan sel darah merah sampai 30%.
Risiko yang terkait dengan splenektomi minimal, dan banyak prosedur sekarang dilakukan dengan laparoskopi. Biasanya, prosedur ditunda bila memungkinkan sampai anak berusia 4-5 tahun atau lebih. Pengobatan agresif dengan antibiotik harus selalu diberikan untuk
setiap keluhan demam sambil menunggu hasil kultur. Dosis rendah Aspirin® setiap hari juga bermanfaat jika platelet meningkat menjadi lebih dari 600.000 / μL pasca splenektomi.
e. Transplantasi sumsum tulang(4)
Transplantasi sumsum tulang untuk talasemia pertama kali dilakukan tahun 1982. Transplantasi sumsum tulang merupakan satu-satunya terapi definitive untuk talasemia. Jarang dilakukan karena mahal dan sulit.
f. Diet talasemia (11)
Pasien dianjurkan menjalani diet normal, dengan suplemen sebagai berikut :
o Vitamin C 100-250 mg/hari selama pemberian kelasi besi.
o Asam Folat 2-5 mg/hari untuk memenuhi kebutuhan yang meningkat.
o Vitamin E 200-400 IU setiap hari.
Sebaiknya zat besi tidak diberikan, dan makanan yang kaya akan zat besi juga dihindari. Kopi dan teh diketahui dapat membantu mengurangi penyerapan zat besi di usus.
10. SKRINNING
Ada 2 pendekatan untuk menghinadari thalassemia:
1. Karena karier thalassemia β bias diketahui dengan mudah, skrinning populasi dan koseling tentang pasangan bisa dilakukan. Bila heterozigot menikah, 1 dari 4 anak mereka bisa menjadi homozigot atau gabungan heterozigot.
2. Bila ibu heterozigot sudah diketahui sebelum lahir, pasangannya bisa diperiksa dan bila termasuk karier, pasangan tersebut ditawari diagnosis prenatal dan terminasi kehamilan pada fetus dengan thalassemia β berat.
Bila populasi tersebut menghendaki pemilihan pasangan, dilakukan skrinning premarital yang bisa dilakukan di sekolah anak. Penting menyediakan program konseling verbal maupun tertulis mengenai skrinning.
Alternatif lain bisa juga dilakukan pemeriksaan terhadap setiap wanita hamil berdasar ras, melalui ukuran eritrosit, kadar Hb A2 (meningkat pada thalassemia-β). Bila kadarnya normal, pasien dikirim ke pusat yang bisa menganalisis rantai α. (4)
11. PROGNOSIS
Prognosis bergantung pada tipe dan tingkat keparahan dari thalassemia. Seperti dijelaskan sebelumnya, kondisi klinis penderita thalassemia sangat bervariasi dari ringan bahkan asimtomatik hingga berat dan mengancam jiwa, tergantung pula pada terapi dan komplikasi yang terjadi. Bayi dengan thalassemia α mayor kebanyakn lahir mati atau lahir hidup dan meninggal dalam beberapa jam. Anak dengan thalassemia dengan transfuse darah biasanya hanya bertahan sampai usia 20 tahun, biasanya meninggal karena penimbunan besi. (9)
BAB III
KESIMPULAN
Thalassemia adalah gangguan pembuatan hemoglobin yang diturunkan. Thalassemia ditemukan tersebar di seluruh ras di Mediterania, Timur Tengah, India sampai Asia Tenggara. Thalassemia memiliki dua tipe utama berdasarkan rantai globin yang hilang pada hemoglobin individu yaitu Thalassemia-α dan thalassemia-β, yang nantinya akan dibagi lagi menjadi beberapa subtipe berdasarkan derajat mutasi (secara genetik) ataupun berat ringannya gejala. Thalassemia diturunkan berdasarkan hukum Mendel, resesif atau ko-dominan. Heterozigot biasanya tanpa gejala, sedangkan homozigot atau gabungan heterozigot gejalanya lebih berat dari thalassemia α dan β. Gejala klinis biasa berupa tanda-tanda anemia seperti pucat, lemah,letih,lesu, tidak aktif beraktifitas atau jarang bermain dengan teman seusianya, sesak nafas kurang konsentrasi, sering pula disertai dengan kesulitan makan, gagal tumbuh, infeksi berulang dan perubahan tulang. Pada pemeriksaan fisik didapatkan facies Cooley, conjungtiva anemis, bentuk tulang yang abnormal, pembesarah lien dan atau hepar. Terapi thalassemia antara lain adalah terapi transfusi, terapi pengikat besi (khelasi), splenektomi, dan transplantasi sumsum tulang. Masing-masing terapi memiliki kriteria dan efek samping tertentu sehingga perlu dipertimbangkan secara seksama. Konseling mengenai thalassemia sangat diperlukan untuk skrining dan pemahaman terhadap penderita. Sampai saat ini, penderita thalassemia yang berat
biasanya tidak dapat bertahan hingga mencapai usia dewasa normal meskipun kemungkinan ini tidak tertutup sama sekali.
DAFTAR PUSTAKA
1. Behrman Richard E., Kliegman Robert, Arvin Ann M., et al. Kelainan Hemoglobin: Sindrom Thalassemia. Ilmu Kesehatan Anak Nelson. Volume 2. Edisi ke-15. Jakarta : Penerbit Buku Kedokteran EGC; 2001. Hal 1708-1712.
2. Yaish Hassan M. Thalassemia. April 30, 2010. Available at : http://emedicine.medscape.com/article/958850- overview .
3. Permono, Bambang H., Sutaryo, Ugrasena, IDG. Sel darah merah: Eritropoisis. Buku Ajar Hematologi- Onkologi Anak. Cetakan ketiga. Ikatan Dokter Indonesia. Jakarta : 2010. Hal 1-6, 16-23.
4. Permono, Bambang H., Sutaryo, Ugrasena, IDG. Hemoglobin Abnormal: Talasemia. Buku Ajar Hematologi- Onkologi Anak.. Cetakan ketiga. Ikatan Dokter Indonesia. Jakarta : 2010. Hal 64-84.
5. Staf Pengajar Ilmu Kesehatan Anak Fakultas Kedokteran Universitas Indonesia. Hematologi. Buku Kuliah Ilmu Kesehatan Anak. Fakultas Kedokteran Universita Indonesia: Bagian Ilmu Kesehatan Anak.
6. U.S Department of Health & Human Services. Thalassemias. Available at:
http://www.nhlbi.nih.gov/health/dci/Diseases/Thalassemia/Thal
assemia_Causes.html.
7. Bleibel, SA. Thalassemia Alpha. August 26, 2009. Available at : http://emedicine.medscape.com/article/206397-overview
8. Takeshita, K. Thalassemia Beta. September 27, 2010. Available at :
http://emedicine.medscape.com/article/206490- overview
9. Yaish Hassan M. Thalassemia: Differential diagnoses & Workup. April
30, 2010. Available at :
http://emedicine.medscape.com/article/958850-diagnosis
10. Hay WW, Levin MJ. Hematologic Disorders. Current Diagnosis and Treatment in Pediatrics. 18th Edition. New York : Lange Medical Books/ McGraw Hill Publishing Division ; 2007. Hal 841-845.
11.Haut, A., Wintrobe MM. The hemoglobinopathies and thalassemias. Forfar and Arneil’s Textbook of Paediatrics. Edisi 7. Chruchill Livingstone. 2010. Hal 1621-1632.