Preformulation in
Solid Dosage Form
A Series of Textbooks and Monographs University of Frankfurt Institute of Pharmaceutical Technology University of Tokyo, Tokyo, Japan
GeoffreyT. Tucker US FDA Center for Drug Evaluation and Research
University of Bradford School of Pharmacy
2. Good Manufacturing Practices for Pharmaceuticals: A Plan for Total Quality Control,Sidney H. Willig, Murray M. Tuckerman, and William S. Hitchings IV
3. Microencapsulation,edited by J. R. Nixon
4. Drug Metabolism: Chemical and Biochemical Aspects,Bernard Testa and Peter Jenner
5. New Drugs: Discovery and Development,edited by Alan A. Rubin 6. Sustained and Controlled Release Drug Delivery Systems,edited by
Joseph R. Robinson
7. Modern Pharmaceutics,edited by Gilbert S. Banker and Christopher T. Rhodes
8. Prescription Drugs in Short Supply: Case Histories,Michael A. Schwartz 9. Activated Charcoal: Antidotal and Other Medical Uses,David O. Cooney 10. Concepts in Drug Metabolism (in two parts),edited by Peter Jenner and
Bernard Testa
11. Pharmaceutical Analysis: Modern Methods (in two parts),edited by James W. Munson
12. Techniques of Solubilization of Drugs,edited by Samuel H. Yalkowsky 13. Orphan Drugs,edited by Fred E. Karch
14. Novel Drug Delivery Systems: Fundamentals, Developmental Concepts, Biomedical Assessments,Yie W. Chien
15. Pharmacokinetics: Second Edition, Revised and Expanded, Milo Gibaldi and Donald Perrier
16. Good Manufacturing Practices for Pharmaceuticals: A Plan for Total Quality Control, Second Edition, Revised and Expanded,Sidney H. Willig,
Murray M. Tuckerman, and William S. Hitchings IV
17. Formulation of Veterinary Dosage Forms,edited by Jack Blodinger 18. Dermatological Formulations: Percutaneous Absorption,Brian W. Barry 19. The Clinical Research Process in the Pharmaceutical Industry,edited by
Gary M. Matoren
20. Microencapsulation and Related Drug Processes,Patrick B. Deasy 21. Drugs and Nutrients: The Interactive Effects,edited by Daphne A. Roe
and T. Colin Campbell
22. Biotechnology of Industrial Antibiotics,Erick J. Vandamme
23. Pharmaceutical Process Validation,edited by Bernard T. Loftus and Robert A. Nash
24. Anticancer and Interferon Agents: Synthesis and Properties, edited by Raphael M. Ottenbrite and George B. Butler
Benjamin J. Gudzinowicz, Burrows T. Younkin, Jr., and Michael J. Gudzinowicz
27. Modern Analysis of Antibiotics,edited by Adjoran Aszalos 28. Solubility and Related Properties,Kenneth C. James
29. Controlled Drug Delivery: Fundamentals and Applications, Second Edition, Revised and Expanded,edited by Joseph R. Robinson and
Vincent H. Lee
30. New Drug Approval Process: Clinical and Regulatory Management, edited by Richard A. Guarino
31. Transdermal Controlled Systemic Medications,edited by Yie W. Chien 32. Drug Delivery Devices: Fundamentals and Applications,edited by
Praveen Tyle
33. Pharmacokinetics: Regulatory•Industrial•Academic Perspectives, edited by Peter G. Welling and Francis L. S. Tse
34. Clinical Drug Trials and Tribulations,edited by Allen E. Cato
35. Transdermal Drug Delivery: Developmental Issues and Research Initiatives, edited by Jonathan Hadgraft and Richard H. Guy
36. Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms, edited by James W. McGinity
37. Pharmaceutical Pelletization Technology,edited by Isaac Ghebre-Sellassie 38. Good Laboratory Practice Regulations,edited by Allen F. Hirsch
39. Nasal Systemic Drug Delivery,Yie W. Chien, Kenneth S. E. Su, and Shyi-Feu Chang
40. Modern Pharmaceutics: Second Edition, Revised and Expanded,edited by Gilbert S. Banker and Christopher T. Rhodes
41. Specialized Drug Delivery Systems: Manufacturing and Production Technology,edited by Praveen Tyle
42. Topical Drug Delivery Formulations,edited by David W. Osborne and Anton H. Amann
43. Drug Stability: Principles and Practices,Jens T. Carstensen 44. Pharmaceutical Statistics: Practical and Clinical Applications,
Second Edition, Revised and Expanded,Sanford Bolton 45. Biodegradable Polymers as Drug Delivery Systems,edited by
Mark Chasin and Robert Langer
46. Preclinical Drug Disposition: A Laboratory Handbook, Francis L. S. Tse and James J. Jaffe
47. HPLC in the Pharmaceutical Industry,edited by Godwin W. Fong and Stanley K. Lam
50. Novel Drug Delivery Systems: Second Edition, Revised and Expanded, Yie W. Chien
51. Managing the Clinical Drug Development Process,David M. Cocchetto and Ronald V. Nardi
52. Good Manufacturing Practices for Pharmaceuticals: A Plan for Total Quality Control, Third Edition,edited by Sidney H. Willig and James R. Stoker 53. Prodrugs: Topical and Ocular Drug Delivery,edited by Kenneth B. Sloan 54. Pharmaceutical Inhalation Aerosol Technology,edited by
Anthony J. Hickey
55. Radiopharmaceuticals: Chemistry and Pharmacology,edited by Adrian D. Nunn
56. New Drug Approval Process: Second Edition, Revised and Expanded, edited by Richard A. Guarino
57. Pharmaceutical Process Validation: Second Edition, Revised and Expanded, edited by Ira R. Berry and Robert A. Nash
58. Ophthalmic Drug Delivery Systems,edited by Ashim K. Mitra 59. Pharmaceutical Skin Penetration Enhancement,edited by
Kenneth A. Walters and Jonathan Hadgraft
60. Colonic Drug Absorption and Metabolism,edited by Peter R. Bieck 61. Pharmaceutical Particulate Carriers: Therapeutic Applications,edited by
Alain Rolland
62. Drug Permeation Enhancement: Theory and Applications,edited by Dean S. Hsieh
63. Glycopeptide Antibiotics,edited by Ramakrishnan Nagarajan
64. Achieving Sterility in Medical and Pharmaceutical Products,Nigel A. Halls 65. Multiparticulate Oral Drug Delivery,edited by Isaac Ghebre-Sellassie 66. Colloidal Drug Delivery Systems,edited by Jo¨rg Kreuter
67. Pharmacokinetics: Regulatory•Industrial•Academic Perspectives, Second Edition,edited by Peter G. Welling and Francis L. S. Tse
68. Drug Stability: Principles and Practices, Second Edition, Revised and Expanded,Jens T. Carstensen
69. Good Laboratory Practice Regulations: Second Edition, Revised and Expanded,edited by Sandy Weinberg
70. Physical Characterization of Pharmaceutical Solids,edited by Harry G. Brittain
71. Pharmaceutical Powder Compaction Technology,edited by Go¨ran Alderborn and Christer Nystro¨m
Simon Benita
74. Oral Mucosal Drug Delivery,edited by Michael J. Rathbone 75. Clinical Research in Pharmaceutical Development,edited by
Barry Bleidt and Michael Montagne
76. The Drug Development Process: Increasing Efficiency and Cost Effectiveness,edited by Peter G. Welling, Louis Lasagna, and Umesh V. Banakar
77. Microparticulate Systems for the Delivery of Proteins and Vaccines,edited by Smadar Cohen and Howard Bernstein
78. Good Manufacturing Practices for Pharmaceuticals: A Plan for Total Quality Control, Fourth Edition, Revised and Expanded,Sidney H. Willig and James R. Stoker
79. Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms: Second Edition, Revised and Expanded,edited by James W. McGinity 80. Pharmaceutical Statistics: Practical and Clinical Applications, Third Edition,
Sanford Bolton
81. Handbook of Pharmaceutical Granulation Technology,edited by Dilip M. Parikh
82. Biotechnology of Antibiotics: Second Edition, Revised and Expanded, edited by William R. Strohl
83. Mechanisms of Transdermal Drug Delivery,edited by Russell O. Potts and Richard H. Guy
84. Pharmaceutical Enzymes,edited by Albert Lauwers and Simon Scharpe´ 85. Development of Biopharmaceutical Parenteral Dosage Forms, edited by
John A. Bontempo
86. Pharmaceutical Project Management,edited by Tony Kennedy
87. Drug Products for Clinical Trials: An International Guide to Formulation• Production•Quality Control,edited by Donald C. Monkhouse and Christopher T. Rhodes
88. Development and Formulation of Veterinary Dosage Forms: Second Edition, Revised and Expanded,edited by Gregory E. Hardee and J. Desmond Baggot
89. Receptor-Based Drug Design,edited by Paul Leff
90. Automation and Validation of Information in Pharmaceutical Processing, edited by Joseph F. deSpautz
91. Dermal Absorption and Toxicity Assessment,edited by Michael S. Roberts and Kenneth A. Walters
92. Pharmaceutical Experimental Design,Gareth A. Lewis, Didier Mathieu, and Roger Phan-Tan-Luu
NMR Spectroscopy,David E. Bugay and W. Paul Findlay
95. Polymorphism in Pharmaceutical Solids,edited by Harry G. Brittain 96. Freeze-Drying/Lyophilization of Pharmaceutical and Biological Products,
edited by Louis Rey and Joan C.May
97. Percutaneous Absorption: Drugs–Cosmetics–Mechanisms–Methodology, Third Edition, Revised and Expanded,edited by Robert L. Bronaugh and Howard I. Maibach
98. Bioadhesive Drug Delivery Systems: Fundamentals, Novel Approaches, and Development,edited by Edith Mathiowitz, Donald E. Chickering III, and Claus-Michael Lehr
99. Protein Formulation and Delivery,edited by Eugene J. McNally 100. New Drug Approval Process: Third Edition, The Global Challenge,
edited by Richard A. Guarino
101. Peptide and Protein Drug Analysis,edited by Ronald E. Reid 102. Transport Processes in Pharmaceutical Systems,edited by
Gordon L. Amidon, Ping I. Lee, and Elizabeth M. Topp 103. Excipient Toxicity and Safety,edited by Myra L. Weiner and
Lois A. Kotkoskie
104. The Clinical Audit in Pharmaceutical Development,edited by Michael R. Hamrell
105. Pharmaceutical Emulsions and Suspensions,edited by Francoise Nielloud and Gilberte Marti-Mestres
106. Oral Drug Absorption: Prediction and Assessment,edited by Jennifer B. Dressman and Hans Lennerna¨s
107. Drug Stability: Principles and Practices, Third Edition, Revised and Expanded,edited by Jens T. Carstensen and C. T. Rhodes 108. Containment in the Pharmaceutical Industry,edited by James P. Wood 109. Good Manufacturing Practices for Pharmaceuticals: A Plan for Total Quality
Control from Manufacturer to Consumer, Fifth Edition, Revised and Expanded,Sidney H. Willig
110. Advanced Pharmaceutical Solids,Jens T. Carstensen 111. Endotoxins: Pyrogens, LAL Testing, and Depyrogenation,
Second Edition, Revised and Expanded,Kevin L. Williams 112. Pharmaceutical Process Engineering,Anthony J. Hickey and
David Ganderton
113. Pharmacogenomics,edited by Werner Kalow, Urs A. Meyer and Rachel F. Tyndale
114. Handbook of Drug Screening,edited by Ramakrishna Seethala and Prabhavathi B. Fernandes
117. Handbook of Pharmaceutical Analysis,edited by Lena Ohannesian and Anthony J. Streeter
118. Pharmaceutical Process Scale-Up,edited by Michael Levin 119. Dermatological and Transdermal Formulations,edited by
Kenneth A. Walters
120. Clinical Drug Trials and Tribulations: Second Edition, Revised and Expanded,edited by Allen Cato, Lynda Sutton, and Allen Cato III 121. Modern Pharmaceutics: Fourth Edition, Revised and Expanded,
edited by Gilbert S. Banker and Christopher T. Rhodes 122. Surfactants and Polymers in Drug Delivery,Martin Malmsten
123. Transdermal Drug Delivery: Second Edition, Revised and Expanded,edited by Richard H. Guy and Jonathan Hadgraft
124. Good Laboratory Practice Regulations: Second Edition, Revised and Expanded,edited by Sandy Weinberg
125. Parenteral Quality Control: Sterility, Pyrogen, Particulate, and Package Integrity Testing: Third Edition, Revised and Expanded,Michael J. Akers, Daniel S. Larrimore, and Dana Morton Guazzo
126. Modified-Release Drug Delivery Technology,edited by Michael J. Rathbone, Jonathan Hadgraft, and Michael S. Roberts
127. Simulation for Designing Clinical Trials:
A Pharmacokinetic-Pharmacodynamic Modeling Perspective, edited by Hui C. Kimko and Stephen B. Duffull
128. Affinity Capillary Electrophoresis in Pharmaceutics and Biopharmaceutics, edited by Reinhard H. H. Neubert and Hans-Hermann Ruttinger€
129. Pharmaceutical Process Validation: An International Third Edition, Revised and Expanded,edited by Robert A. Nash and Alfred H. Wachter
130. Ophthalmic Drug Delivery Systems: Second Edition, Revised and Expanded,edited by Ashim K. Mitra
131. Pharmaceutical Gene Delivery Systems,edited by Alain Rolland and Sean M. Sullivan
132. Biomarkers in Clinical Drug Development,edited by John C. Bloom and Robert A. Dean
133. Pharmaceutical Extrusion Technology,edited by Isaac Ghebre-Sellassie and Charles Martin
134. Pharmaceutical Inhalation Aerosol Technology: Second Edition, Revised and Expanded,edited by Anthony J. Hickey
135. Pharmaceutical Statistics: Practical and Clinical Applications, Fourth Edition, Sanford Bolton and Charles Bon
Second Edition, Revised and Expanded,edited by Louis Rey and Joan C. May
138. Supercritical Fluid Technology for Drug Product Development, edited by Peter York,Uday B. Kompella, and Boris Y. Shekunov
139. New Drug Approval Process: Fourth Edition, Accelerating Global Registrations,edited by Richard A. Guarino
140. Microbial Contamination Control in Parenteral Manufacturing, edited by Kevin L. Williams
141. New Drug Development: Regulatory Paradigms for Clinical Pharmacology and Biopharmaceutics,edited by Chandrahas G. Sahajwalla
142. Microbial Contamination Control in the Pharmaceutical Industry, edited by Luis Jimenez
143. Generic Drug Product Development: Solid Oral Dosage Forms, edited by Leon Shargel and Izzy Kanfer
144. Introduction to the Pharmaceutical Regulatory Process,edited by Ira R. Berry
145. Drug Delivery to the Oral Cavity: Molecules to Market,edited by Tapash K. Ghosh and William R. Pfister
146. Good Design Practices for GMP Pharmaceutical Facilities,edited by Andrew Signore and Terry Jacobs
147. Drug Products for Clinical Trials, Second Edition,edited by Donald Monkhouse, Charles Carney, and Jim Clark 148. Polymeric Drug Delivery Systems,edited by Glen S. Kwon 149. Injectable Dispersed Systems: Formulation, Processing, and
Performance,edited by Diane J. Burgess
150. Laboratory Auditing for Quality and Regulatory Compliance,Donald Singer, Raluca-Ioana Stefan, and Jacobus van Staden
151. Active Pharmaceutical Ingredients: Development, Manufacturing, and Regulation,edited by Stanley Nusim
152. Preclinical Drug Development,edited by Mark C. Rogge and David R. Taft 153. Pharmaceutical Stress Testing: Predicting Drug Degradation,
edited by Steven W. Baertschi
154. Handbook of Pharmaceutical Granulation Technology: Second Edition,edited by Dilip M. Parikh
155. Percutaneous Absorption: Drugs–Cosmetics–Mechanisms–Methodology, Fourth Edition,edited by Robert L. Bronaugh and Howard I. Maibach 156. Pharmacogenomics: Second Edition,edited by Werner Kalow,
Urs A. Meyer and Rachel F. Tyndale
157. Pharmaceutical Process Scale-Up, Second Edition,edited by Michael Levin 158. Microencapsulation: Methods and Industrial Applications, Second Edition,
Ram B. Gupta and Uday B. Kompella
160. Spectroscopy of Pharmaceutical Solids,edited by Harry G. Brittain 161. Dose Optimization in Drug Development,edited by Rajesh Krishna 162. Herbal Supplements-Drug Interactions: Scientific and
Regulatory Perspectives,edited by Y. W. Francis Lam, Shiew-Mei Huang, and Stephen D. Hall
163. Pharmaceutical Photostability and Stabilization Technology, edited by Joseph T.Piechocki and Karl Thoma
164. Environmental Monitoring for Cleanrooms and Controlled Environments, edited by Anne Marie Dixon
165. Pharmaceutical Product Development: In Vitro-In Vivo Correlation,edited by Dakshina Murthy Chilukuri, Gangadhar Sunkara, and David Young 166. Nanoparticulate Drug Delivery Systems,edited by Deepak Thassu,
Michel Deleers, and Yashwant Pathak
167. Endotoxins: Pyrogens, LAL Testing and Depyrogenation, Third Edition,edited by Kevin L. Williams
168. Good Laboratory Practice Regulations, Fourth Edition, edited by Anne Sandy Weinberg
169. Good Manufacturing Practices for Pharmaceuticals, Sixth Edition,edited by Joseph D. Nally
170. Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-soluble Drugs,edited by David J. Hauss 171. Handbook of Bioequivalence Testing,Sarfaraz K. Niazi
172. Advanced Drug Formulation Design to Optimize Therapeutic Outcomes, edited by Robert O. Williams III, David R. Taft, and Jason T. McConville 173. Clean-in-Place for Biopharmaceutical Processes,edited by
Dale A. Seiberling
174. Filtration and Purification in the Biopharmaceutical Industry, Second Edition, edited by Maik W. Jornitz and Theodore H. Meltzer
175. Protein Formulation and Delivery, Second Edition,edited by Eugene J. McNally and Jayne E. Hastedt
176. Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms,edited by James McGinity and Linda A. Felton
177. Dermal Absorption and Toxicity Assessment, Second Edition, edited by Michael S. Roberts and Kenneth A. Walters
Edited by
Moji Christianah Adeyeye
Duquesne University Pittsburgh, Pennsylvania, USAHarry G. Brittain
Center for Pharmaceutical PhysicsMilford, New Jersey, USA
Preformulation in
Solid Dosage Form
52 Vanderbilt Avenue New York, NY 10017
ª2008 by Informa Healthcare USA, Inc. Informa Healthcare is an Informa business
No claim to original U.S. Government works
Printed in the United States of America on acid-free paper 10 9 8 7 6 5 4 3 2 1
International Standard Book Number-10: 0-8247-5809-9 (Hardcover) International Standard Book Number-13: 978-0-8247-5809-7 (Hardcover)
This book contains information obtained from authentic and highly regarded sources. Reprinted material is quoted with permission, and sources are indicated. A wide variety of references are listed. Reasonable efforts have been made to publish reliable data and information, but the author and the publisher cannot assume responsibility for the validity of all materials or for the consequence of their use.
No part of this book may be reprinted, reproduced, transmitted, or utilized in any form by any electronic, mechanical, or other means, now known or hereafter invented, including photocopying, microfilming, and recording, or in any information storage or retrieval system, without written permission from the publishers.
For permission to photocopy or use material electronically from this work, please access www. copyright.com (http://www.copyright.com/) or contact the Copyright Clearance Center, Inc. (CCC) 222 Rosewood Drive, Danvers, MA 01923, 978-750-8400. CCC is a not-for-profit organization that provides licenses and registration for a variety of users. For organizations that have been granted a photocopy license by the CCC, a separate system of payment has been arranged.
Trademark Notice:Product or corporate names may be trademarks or registered trademarks, and are used only for identification and explanation without intent to infringe.
Library of Congress Cataloging-in-Publication Data
Preformulation in solid dosage form development / edited by Moji Christianah Adeyeye, Harry G. Brittain.
p. ; cm. – (Drugs and the pharmaceutical sciences ; 178) Includes bibliographical references and index.
ISBN-13: 978-0-8247-5809-7 (hb : alk. paper) ISBN-10: 0-8247-5809-9 (hb : alk. paper)
1. Solid dosage forms. 2. Drug development. 3. Pharmaceutical chemistry. I. Adeyeye, Moji C. II. Brittain, H. G. III. Series.
[DNLM: 1. Chemistry, Pharmaceutical. 2. Dosage Forms. 3. Pharmaceutical Preparations–chemical synthesis. W1 DR893B v.178 2008 / QV 744 P923 2008]
RS201.S57P74 2008
6150.19–dc22 2007033181
For Corporate Sales and Reprint Permissions call 212-520-2700 or write to: Sales Department, 52 Vanderbilt Ave., 16th floor, New York, NY 10017.
Visit the Informa web site at www.informa.com
Prefaces
I
The conception of writing a book on preformulation began many years ago after noticing the dearth of information on characterization of chemical entities and excipients in pharmaceutical drug development while developing lecture notes for the Formulation and Development course that I teach in my school. As a teacher in the “ivory tower,” I found this intriguing, knowing that future industry scientists passing through institutions of higher learning need exposure to the basic information as well as current and relevant advances in conducting preformulation research in the industry. During the 1996 American Association of Pharmaceutical Scientists meeting, I brought this up at the preformulation focus group meeting where several colleagues volunteered to contribute chapters toward the making of the book.
The intent of this book is to equip both academia and the pharmaceutical industry with adequate basic and applied principles important to the characterization of drugs, excipients, and products during the preformulation stage in development. The issues of predictability, identification, and product development of drug substances during the preformulation phase and into Phase I clinical trials have been addressed. The relevance of these to setting acceptance criteria (indicative of building quality into the product instead of adding quality) for regulatory purposes has been emphasized. Each chapter has been written with the industry scientist/practitioner and the regulatory authorities in mind so that the book can serve as a handbook and reference text for answering questions or problem solving.
During the evolution of the writing process, several colleagues encouraged me and contributed toward the rationale for writing the book, and these include George Wong and Ivan Santos. I am very grateful for my co-editor, Harry G. Brittain, for his ebullience and encouragement during the making of the book.
Moji Christianah Adeyeye
II
If you ask 20 workers in the pharmaceutical field what is meant by the science of “preformulation,” I have no doubt that you will receive 20 different answers. To the early-stage scientist, it represents the process of characterizing a new drug substance, to learn about its properties and tendencies. Working through a pre-planned program of investigational work, scientists obtain a wide range of information. Clearly they cannot learn everything, and in fact, the time and money constraints of modern drug development often dictate that the minimal amount of work is done that will satisfy a regulatory agency. Industrial preformulation groups march through two or three compounds per month, gather the type of data they think will be of use later on, and write a report that they hope will be read by everyone who comes after them in the process.
To the latter-stage scientist, preformulation represents the stage where someone else has profiled a drug candidate to such a degree that he or she has all the information needed to complete the development process. Unfortunately, since this worker has usually had relatively little input into the design of the preformulation studies, it is not a given that the preformulation report will contain all that is really needed. There ordinarily follows a scrambling for new information, or (worse yet) an era of trouble shooting and formulation fixing. Everyone in late-stage development under-stands that the process is inevitable, but no one has to like it.
In the present volume, Professor Adeyeye and I have tried to outline a program of preformulation that fits better with the modern school of drug development. All of the traditional investigations are contained, but we have tried to look forward through the development process so that appropriate preformulation activities are included in the early work. We equally believe that computational prediction can play an important role in the characterization of drug compounds, and have used the term “preliminary preformulation” to cover such work. It is our hope that we have covered all the topics of importance to preformulation work, and that we have set out a program that can be successfully applied to the majority of drug candidates.
Contents
Prefaces iii Contributors vii
PART 1: INTRODUCTION
1.1 Introduction and Overview to the Preformulation Development of Solid Dosage Forms 1
Harry G. Brittain
PART 2: PRELIMINARY PREFORMULATION
2.1 Accelerating the Course of Preliminary Preformulation Through Prediction of Molecular Physical Properties and Integrated Analytical Data Management 17
Robert S. DeWitte, Michel Hachey, and Harry G. Brittain
2.2 Prediction of Crystallographic Characteristics 41
Stephen Spanton
2.3 Salt Selection for Pharmaceutical Compounds 63
Sherif I. Badawy, Miriam K. Franchini, and Munir A. Hussain
2.4 Intelligent Preformulation Design and Predictions Using Artificial Neural Networks 81
Nkere K. Ebube
PART 3: PROFILING THE DRUG SUBSTANCE
3.1 Developing a Profile of the Active Pharmaceutical Ingredient 115
Harry G. Brittain
3.2 Particle Morphology and Characterization in Preformulation 145
Alan F. Rawle
3.3 Preparation and Identification of Polymorphs and Solvatomorphs 185
Harry G. Brittain
3.4 X-Ray Diffraction Methods for the Characterization of Solid Pharmaceutical Materials 229
J. R. Blache´re and Harry G. Brittain
3.5 Spectroscopic Methods for the Characterization of Drug Substances 253
Harry G. Brittain
3.6 Thermal Analysis and Calorimetric Methods for the Characterization of New Crystal Forms 279
Denette K. Murphy and Shelley Rabel
3.7 Solubility Methods for the Characterization of New Crystal Forms 323
Harry G. Brittain
PART 4: DEVELOPMENT OF THE IDEAL FORMULATION
4.1 Overview of the Solid Dosage Form Preformulation Program 347
Harry G. Brittain
4.2 Drug–Excipient Interaction Occurrences During Solid Dosage Form Development 357
Moji Christianah Adeyeye
4.3 Methodology for the Evaluation of Chemical and Physical Interactions Between Drug Substances and Excipients 437
Harry G. Brittain
4.4 Dissolution Testing 477
George Wong and Charles C. Collins
PART 5: BEYOND PERFORMULATION
5.1 Structure, Content, and Format of the Preformulation Report 557
Ram N. Gidwani
5.2 Significance of Drug Substance Physicochemical Properties in Regulatory Quality by Design 571
Sau Lawrence Lee, Andre S. Raw, and Lawrence Yu
Contributors
Moji Christianah Adeyeye School of Pharmacy, Duquesne University, Pittsburgh, Pennsylvania, U.S.A.
Sherif I. Badawy Bristol-Myers Squibb Pharmaceutical Research Institute, New Brunswick, New Jersey, U.S.A.
J. R. Blache´re Department of Materials Science and Engineering, University of Pittsburgh, Pittsburgh, Pennsylvania, U.S.A.
Harry G. Brittain Center for Pharmaceutical Physics, Milford, New Jersey, U.S.A.
Charles C. Collins College of Pharmacy, East Tennessee State University, Johnson City, Tennessee, U.S.A.
Robert S. DeWitte Advanced Chemistry Development, Inc., Toronto, Ontario, Canada
Nkere K. Ebube Biovail Technologies, Chantilly, Virginia, U.S.A.
Miriam K. Franchini Sanofi-Aventis, Bridgewater, New Jersey, U.S.A.
Ram N. Gidwani Pharmaceutical Consultant, Milford, New Jersey, U.S.A.
Michel Hachey Advanced Chemistry Development, Inc., Toronto, Ontario, Canada
Munir A. Hussain Bristol-Myers Squibb Pharmaceutical Research Institute, New Brunswick, New Jersey, U.S.A.
Sau Lawrence Lee Office of Generic Drugs, United States Food and Drug Administration, Rockville, Maryland, U.S.A.
Denette K. Murphy Bristol-Myers Squibb Company, New Brunswick, New Jersey, U.S.A.
Shelley Rabel ALZA Corporation, Mountain View, California, U.S.A.
Andre S. Raw Office of Generic Drugs, United States Food and Drug Administration, Rockville, Maryland, U.S.A.
Alan F. Rawle Malvern Instruments, Westborough, Massachusetts, U.S.A.
Stephen Spanton GPRD Structural Chemistry, Abbott Laboratories, Abbott Park, Illinois, U.S.A.
George Wong Global R&D Operations, Johnson & Johnson, Skillman, New Jersey, U.S.A.
Part 1: Introduction
1.1
Introduction and Overview to the
Preformulation Development of Solid
Dosage Forms
Harry G. Brittain
Center for Pharmaceutical Physics, Milford, New Jersey, U.S.A.
Once an organic compound has been discovered and shown by one scientific group to have some type of desirable pharmacological activity, it will remain a mere curiosity unless a completely different group of scientists incorporate the compound in a formulation that facilitates its activity. This latter group of investigators constitutes the development effort, and it is their work that turns an interesting compound into an actual drug substance.
Out of the many types of formulations that could be contemplated for a drug substance, the solid oral dose form (i.e., tablets and capsules) con-tinues to be the most important. For this reason, the remainder of the coverage in this volume will center on the development of solid dosage forms, although many of the principles involved will be seen to be applicable to other dosage forms such as liquids, suspensions, emulsions, semisolids, suppositories, and aerosols, to name a few.
Historically, the preformulation stage of drug development has been considered to consist mostly of drug substance profiling, and indeed most of the leading reviews have followed this view (1–6). However, there is far more to the development of drug products than characterization of the active pharmaceutical ingredient, and in its fullest incarnation, preformulation would also extend to studies of drug–excipient compatibilities. At this point, one could simply employ the Edisonian approach of trying all combinations of drugs and excipients to see what would constitute an appropriate for-mulation, a method that has come to be known today as high-throughput
screening. Clearly, it makes more sense to develop profiles of the physical and chemical properties of drug substances and excipients, and to then use this information in order to develop more robust formulations.
For example, while the degree of acidity or basicity of a substance dissolved in an aqueous medium can be adequately defined in terms of pH, comparable expressions for the acidity or basicity of the surface of a solid are more complicated. Such evaluations would be of importance to a pre-formulation study, as it is known that acidic or basic surfaces of solids can function as catalytic agents. The a priori knowledge regarding the degree of acidity of a surface, or the stoichiometry of binding sites, would be of great use to formulators, and its acquisition would serve to streamline pre-formulation studies, especially if the body of information was available for the drug substance as well as the excipients intended for its formulation. In the absence of such knowledge, formulators can only assume the existence of certain characteristics of the materials they are working with, and then base their compounding on assumptions rather than hard facts.
The depth of the problem is extremely important, since in a dosage form, under suitable conditions, a drug substance may undergo a variety of inter-actions or transformations through one or more chemical or physical reac-tions. The understanding of these solid–solid interactions is highly critical to a successful outcome for the development process, as they can lead to the for-mation of new impurities, incomplete mass balance, destruction of the dosage form, and changes in physicochemical properties (stability, solubility, dis-solution profile, degree of crystallinity, and hygroscopicity). Any or all of these could have a most unfortunate outcome that ultimately results in an inability to obtain successful registration of the drug substance. Hence, the elucidation of possible chemical and/or physical reactions is an absolute requirement to establish the stability of a given dosage form, and an under-standing of the plausible range of solid–solid reactions available to a drug substance should be established once a full preformulation study is completed. After careful evaluation of the current trends and practices in pre-formulation, and considering where the field ought to be headed, it seemed appropriate to define four areas of activity. These areas roughly follow the chronological sequence of drug development and can be envisioned as beginning with a preliminary preformulation stage, continuing with a pro-gram of drug substance profiling and development of the ideal formulation, and concluding with technology transfer steps.
Now that the sophistication of computational programs is reaching the point that one may calculate many of these quantities to an acceptable degree of accuracy, one may use the structural formula of a compound to calculate its ionization constant (S), the pH dependence of its partition coefficient, and its aqueous solubility tendencies. Very often, this informa-tion alone can be used to determine whether the free acid or free base form of the compound will have sufficient properties or whether a salt form of the substance might be more appropriate.
As an example, consider ibuprofen (2-(4-isobutylphenyl)propanoic acid) as a potential drug substance. The structure of the molecule is fairly simple (given below), and one would anticipate the important physical chemistry to be dominated by the chemistry of the carboxylate group. Using the PhysChem program [sourced from Advanced Chemistry Development (ACD)], the pKa value of the carboxylate group can be calculated to be 4.41 –0.01. Although this pKavalue is not exactly equal to the reported ionization constant that was determined to be in the range of 4.5 to 4.6 by potentiometric titration in mixed organic/aqueous solvent systems (7), the number is sufficiently close to reality so that it may be used to deduce other important properties of the molecule.
O
OH CH3
C H3
CH3
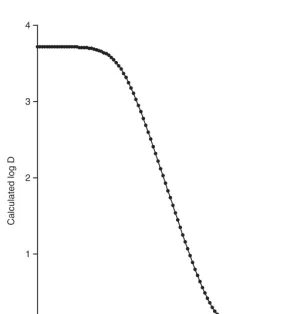
The next step in the computational process would be to calculate the partition coefficient and the pH dependence of the distribution coefficient. For ibuprofen, the ACD program calculates the octanol/water partition coefficient to be log P¼3.72– 0.23, indicating that the compound is expected to be substantially hydrophobic in its neutral form. As might be anticipated, the carboxylate group of ibuprofen yields a strong pH depen-dence in the distribution coefficient, as shown in Figure 1. The calculation indicates that while ibuprofen is predicted to be hydrophobic at low pH, it is also predicted to be hydrophilic at high pH once the carboxylate group becomes ionized.
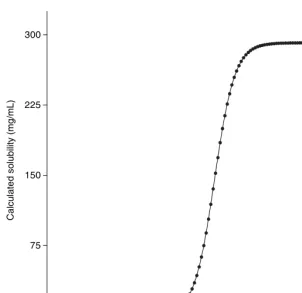
It is a fundamental precept of solubility theory that hydrophilic compounds should exhibit good solubility in aqueous media. As shown in Figure 2, the calculated pH dependence of solubility is consistent with this prediction, with the compound being predicted to be highly soluble above pH 7.
maximize its aqueous solubility. If this decision was made, then the next step would entail prediction of an appropriate salt form. Fortunately, the ioni-zation constants for potential salt formers can be calculated with relative ease. It is relatively easy to show that for the reaction of ibuprofen free acid (H-ibu) with a basic substance (B) to yield the salt (HB+)(ibu ),
H-ibuþBÐHBþþibu ð1Þ
the value for the salt formation constant (Ks),
Ks¼½ibu ½HB
þ
½H-ibu½B ð2Þ
is given by (8)
Ks¼KaKb=Kw ð3Þ
2 4 6 8 10 12 14
0 1 2 3 4
pH
Calculated log D
Figure 1 pH dependence of the distribution coefficient of ibuprofen, calculated
In Equation 3, Ka is the ionization constant expression for the free acid,Kbis the ionization constant expression for the free base, andKwis the autoionization constant of water. When converted to the Sørensen scale, Eq. 3 becomes
pKs¼pKaþpKb pKw ð4Þ
The value of Equations 3 and 4 becomes evident when we see that they can be used to make rapid deductions regarding the strength of a particular salt species. Suppose one were contemplating forming a salt between ibu-profen (whose pKavalue was calculated to be 4.41) and a base having a pKa value of 9.45: For the base, it would follow that the pKbwould equal 4.55, and since pKwequals 14.0 at 25C, the pKsof the salt would equal 5.04 and Ks would equal approximately 109,650. A reaction characterized by an equilibrium constant of this magnitude would clearly go to completion, and one would predict that the salt in question would be formed without diffi-culty. Once the range of acceptable acidic salt formers has been determined,
2 4 6 8 10 12 14
0 75 150 225 300
pH
Calc
u
lated sol
u
bility (mg/mL)
Figure 2 pH dependence of the aqueous solubility of ibuprofen, calculated using the
one only needs to consult the various compilations of pharmaceutically acceptable acids (9,10) to specify the list of salts that would be actually prepared in the laboratory.
The ability to calculate log(Ks) values can be of great value in designing the scope of a salt-selection study in that it can be used to select potential salt-forming agents solely on the basis of their ionization constant values. If one accepts the definition of an appropriate salt as one whose degree of formation equals 99% or higher, then one would only attempt to form ibuprofen salts with bases whose pKavalues were sufficiently high, so as to guarantee the required degree of reaction. Assuming a 1:1 stoichio-metry, one may use the quadratic equation to solve for the degree of dis-sociation associated with salts having variousKsvalues. The equations have been solved analytically, and as shown in Figure 3, for ibuprofen, one finds that the critical pK exceeds 8.41. For example, the formation of sodium or potassium salts (for which pKais approximately 14) is obvious, as would be the formation of salts with arginine (pKa¼9.59), lysine (pKa¼9.48), ethanolamine (pKa¼9.16), diethanolamine (pKa¼8.71), and erbumine (pKa¼10.68).
7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 94
95 96 97 98
99 100
pKa of salt-forming base
P
e
rcentage of salt f
o
rmed
Figure 3 Degree of salt formation calculated for the reaction of ibuprofen with
At this point in the preformulation process, it becomes necessary to go into the laboratory and begin to perform the experimentation that will ultimately lead to the selection of the drug candidate in its proper solid-state form. To illustrate the process, we will continue with the ibuprofen example, where it has already been established that the formation of a salt form of the drug substance should lead to enhanced solubility and hence dissolution rate. Ibuprofen represents an interesting situation in that it contains one center of dissymmetry, and hence the free acid is capable of being obtained either as a racemic mixture or as one of the separated enantiomers. The physical properties of these forms are quite different, exhibiting different crystal structures, melting points, and degrees of aqueous solubility (7).
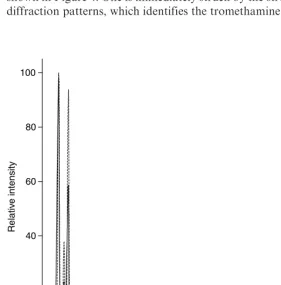
Ibuprofen readily forms salts with tromethamine (2-amino-2-(hydro-xymethyl)propane-1,3-diol), and the X-ray powder diffraction patterns of the tromethamine:(S)-ibuprofen and tromethamine:(RS)-ibuprofen salts are shown in Figure 4. One is immediately struck by the strong similarities in the diffraction patterns, which identifies the tromethamine salts as belonging to
5 10 15 20 25 30 35 40
0 20 40 60
80 100
Scattering angle (degrees 2θ)
Relativ
e
intensity
Figure 4 X-ray powder diffraction patterns of the tromethamine salts with
a conglomerate system. In a conglomerate, the different enantiomers of the solid substance crystallize in different crystals, so that the observed dif-fraction patterns of the separated enantiomers and the racemic mixture are the same (11).
Optical photomicrographs of the tromethamine salts with (S)-ibuprofen and (RS)-ibuprofen are shown in Figure 5. It was found that the particles of both salts were obtained in the form of thin flat plates, but the edges were poorly defined. The crystals of both salts exhibited strong birefringence, which would be consistent with the high degree of crystallinity observed in the X-ray diffraction studies.
Figure 5 Optical photomicrographs of the tromethamine salts with (S)-ibuprofen
The structural equivalence (i.e., conglomerate nature) of the tro-methamine salts with (S)-ibuprofen and (RS)-ibuprofen is further indicated in the equivalence of their other physical properties. The differential scan-ning calorimetry thermograms of the salts (Fig. 6) are effectively the same, with the (S)-ibuprofen salt exhibiting a melting endotherm at 158.4C
(enthalpy of fusion equal to 159.1 J/g) and the (RS)-ibuprofen salt having a melting endotherm of 158.9C (enthalpy of fusion equal to 160.2 J/g). It is
to be noted that these fusion characteristics represent a vast improvement over those of the parent free acids, where (S)-ibuprofen was found to exhibit a melting endotherm at 58.8C (enthalpy of fusion equal to 67.9 J/g) and
(RS)-ibuprofen exhibited a melting endotherm of 77.3C (enthalpy of fusion
equal to 77.9 J/g).
Since the crystal structures of the (S)-ibuprofen:tromethamine and (RS)-ibuprofen:tromethamine salts are effectively the same, one would anticipate that the respective solid-state spectra would also be equivalent. This is evident in the fingerprint region of the infrared absorption spectra in Figure 7 and in the Raman spectra in Figure 8. It is clear that the
130 140 150 160 170
–15.0 –12.5 –10.0 –7.5 –5.0 –2.5 0.0
Temperature (°C)
Heat flo
w
Figure 6 Differential scanning calorimetry thermograms of the tromethamine salts
tromethamine salt prepared from the enantiomerically pure and racemic free acids could not be differentiated on the basis of their solid-state spectra.
Although the frequencies of individual group vibrations as measured using either infrared absorption spectroscopy or Raman scattering must always be the same for a given compound, the difference in the nature of the selection rules governing the infrared absorption and Raman scattering processes often leads to differences in the relative intensities, which makes the respective spectra appear to be different (12). This has been illustrated in Figure 9, where one may note the apparent nonequivalence of the infrared absorption spectrum and the Raman spectrum of the (S )-ibuprofen:tro-methamine salt. Generally, symmetric vibrations will have the most intensity in a Raman spectrum, and the nonsymmetric vibrations will tend to tolerate the infrared absorption spectrum. Hence, it is prudent to obtain both types of vibrational spectra for a compound under development in order to build up a database of characteristics that may prove to be useful during the study of drug–excipient interactions.
600 800 1000 1200 1400 1600 1800 0.00
0.05 0.10 0.15 0.20 0.25 0.30 0.35
Energy (cm–1)
Absorbance
Figure 7 Infrared absorption spectra of the tromethamine salts with (S)-ibuprofen
The solubility of (RS)-ibuprofen free acid has been reported to be less than 0.1 mg/mL, but the aqueous solubilities of the tromethamine salts with (RS)-ibuprofen and (S)-ibuprofen were found to exceed 100 mg/mL. Given their high degree of solubility in water, and their superior physical char-acteristics (as evidenced by the thermal analysis results), it follows that if one were given a choice, one would naturally choose to develop one of the tromethamine salts rather than the free acid.
Once the form of the drug substance has been chosen, the next stage in the preformulation program would entail the study of how the compound interacts with excipients of interest. Excipients are naturally chosen on their functional basis, properties that are essential to the release of the drug substance from the dosage form. For a solid dosage form designed to rapidly release the drug substance upon contact with a fluid, one normally seeks a formulation where there are no physical or chemical interactions between the various components. For solid dosage forms, an interaction usually is considered to represent a bad situation, since the quality and stability of the formulation will ordinarily be adversely affected.
250 450 650 850 1050 1250 1450 1650 0
20 40 60
80 100
Raman shift (cm–1)
Intensity
Figure 8 Raman spectra of the tromethamine salts with (S)-ibuprofen (solid trace)
A number of reviews have been published that cover the scope of potential solid-state reactions, but in general, one may find what one needs to know in Refs. 13 and 14 and in the other chapters in this book.
One convenient classification for solid-state reactions specifies the natures of the phases involved. For example, the reactions between solids and gases may involve the interaction of the solid substance with water vapor (either sorption or desorption), or with oxygen or other gases. The solid–gas reaction may result in a phase transformation, typically causing the formation of a hydrate or a solvate crystal form. Reactions between solids and liquids can result in hydrolysis of the drug substance [see, for example, the work reported in Ref. 15 on aspirin formulations] or in other acid–base chemistry, or in extreme cases, they may result in the solubiliza-tion of the components involved. One must always monitor whether a phase transformation takes place when a liquid interacts with a solid, as solution-mediated phase transformations are well known.
200 400 600 800 1000 1200 1400 1600 1800 0
10 20 30 40 50 60 70
80 90 100
Energy (cm–1)
Intensity
Figure 9 Infrared absorption spectrum (solid trace) and Raman spectrum (dashed
There are also a number of solid–solid reactions that are possible. Among these are association processes, such as those observed in disper-sions of indomethacin and polyvinylpyrrolidone (16). One may encounter adsorption reactions, as was noted in the adsorption of ketotifen on Ac-Di-Sol (17). Drug substances may become solubilized in one of the other components, demonstrated, for example, by the existence of a complete phase diagram for the quinine–phenobarbital system (18). The well-known Maillard reaction of lactose with amines can be typified by the addition reaction of fluoxetine with spray-dried lactose (19). Desolvation reactions are frequently studied using spectroscopic techniques, such as the use of near-infrared spectroscopy to monitor the loss of isopropyl alcohol from loracarbef solvatomorphs (20). There are many physical and chemical techniques suitable for the study of drug decomposition in formulations, but one of the more creative methods entails the correlation of quantitative color parameters with and drug content in formulations of flucloxin (21). Of course, one must always remember the possibility of phase transformations in solids, and the number of solid-state phase changes that have been published is very large.
To evaluate the compatibility between drug substance and excipients, one will blend the API with excipients at levels that are realistic with respect to the proposed formulation. For example, if a lubricant is to be used at the 0.5% level in a formulation, the evaluation of a 50% substance–excipient mixture would not be appropriate. Each blend is then stored under an appropriate set of accelerated stress conditions (typically elevated tem-perature and humidity) and subsequently tested for drug substance quality after an appropriate equilibration period.
The purity and impurity profiling is ordinarily conducted using a chromatographic procedure, while the physical characteristics are normally followed by means of the appropriate physical analysis methodology. The methodologies suitable for the study of chemical compatibilities include high-performance liquid chromatography (HPLC), capillary electrophor-esis, gas chromatography (GC), mass spectrometry (especially coupled with GC or HPLC), nuclear magnetic resonance, and vibrational spec-troscopy. The methodologies suitable for the study of physical compat-ibilities include microscopy (either optical or scanning electron), X-ray powder diffraction, thermal analysis (typically differential scanning calorimetry and thermogravimetry), ultraviolet/visible or near-infrared diffuse reflectance spectroscopy, solid-state nuclear magnetic resonance, and solid-state vibrational spectroscopy. The technology to be used during this phase is determined by the nature of the problem, but ordinarily, the correct approach entails a multidisciplinary use of methodology where the optimal tools are selected to obtain the information of interest.
and Phase 2 clinical studies. These formulations should not only suit their intended purpose, but also lead the way to the eventual Phase 3 formulation. Once a few trial formulations of the drug substance have been devel-oped, and tablets or capsules prepared from these, dissolution testing can be taken up in the evaluation studies. Most studies are conducted using the batch dissolution method, where the analyzed concentration of a well-stirred solution is taken as being representative of the entire volume of the dis-solution medium. In a disdis-solution profile of a good formulation, the analyte concentration will increase from its initial zero value until a limiting con-centration is attained that indicates full dissolution of the drug substance.
A simple use of dissolution testing as a means of formulation optimi-zation is shown in Figure 10, which contains dissolution profiles of two pro-totype tablet formulations prepared using the (S)-ibuprofen:tromethamine salt as the drug substance. It was found that only partial dissolution of the
0 5 10 15 20 25 30
0 20 40 60
80 100
Time (minutes)
P
e
rcent dissolv
ed
Figure 10 Dissolution profiles of various prototype tablet formulations prepared
salt could be obtained for a formulation consisting of 25% API and 75% microcrystalline cellulose, but that full dissolution could be obtained from a formulation consisting of 25% API, 65% microcrystalline cellulose, and 10% sodium starch glycolate. These observations seem to indicate not only the existence of a drug–excipient interaction between the (S )-ibuprofen:tro-methamine salt and the microcrystalline cellulose, but also that the inclusion of a disintegrant in the formulation can serve to mitigate this effect.
At the conclusion of the second phase of the preformulation work, one should have developed a catalog of the plausible solid-state reactions accessible to the drug substance. Any concerns that developed during this work should receive a degree of attention that is roughly proportional to their likelihood of actually being encountered, making it essential that the studies are performed at appropriate concentration levels. Clearly, since the scope of possible solid–solid reactions encompasses both chemical and physical interactions, it follows that both types need to be studied by an appropriate methodology. Although real-time data would be preferable, in the absence of such data, the use of accelerated studies can greatly expedite matters.
No matter how good or thorough the preformulation program has been, there is no doubt that the work is wasted unless the technology can be transferred to the next stage. This accumulated body of knowledge will enable formulation scientists to build upon what was learned, and will enable them to use the catalog of drug–excipient interactions to avoid additional formulation changes doomed to failure. The physical and the chemical methods of characterization developed during preformulation will be of equal interest to the analytical scientists, who generally use this information to optimize existing methods and develop new ones.
Every organization develops its own models for this technology-transfer process, but there is usually no better method than to collect everything that was learned in one or more preformulation reports. Such documentation may even be filed with a new drug application at some point, should that possibility develop.
The various topics outlined in this brief introduction and overview of the preformulation development of solid dosage forms will be expounded in the following chapters.
REFERENCES
1. Fiese EF, Hagen TA. Preformulation (chap. 8). In: Lachman L, Lieberman HA, Kanig JL, eds. Theory and Practice of Industrial Pharmacy. Philadelphia: Lea & Febiger, 1986, pp. 171–96.
3. Wadke DA, Serajuddin ATM, Jacobson H. Preformulation testing (chap. 1). In: Lieberman HA, Lachman L, Schwartz JB, eds. Pharmaceutical Dosage Forms, 2nd ed., vol. 1. New York: Marcel Dekker, 1989, pp. 1–73.
4. Carstensen JT. Preformulation (chap. 9). In: Carstensen JT, Rhodes CT, eds. Drug Stability, 3rd ed. New York: Marcel Dekker, 2000, pp. 237–60. 5. Carstensen JT. Preformulation (chap. 7). In: Banker GS, Rhodes CT, eds.
Modern Pharmaceutics, 4th ed. New York: Marcel Dekker, 2002, pp. 167–85. 6. Ando HY, Radebaugh GW. Property-based drug design and preformulation (chap. 38). In: Hendrickson R, ed. Remington: The Science and Practice of Pharmacy, 21st edn. Philadelphia: Lippincott Williams & Wilkins, 2005, pp. 720–44.
7. Higgins JD, Gilmor TP, Martellucci SA, Bruce RD, Brittain HG. Ibuprofen (chap. 6). In: HG Brittain, ed. Analytical Profiles of Drug Substances and Excipients, vol. 27, San Diego: Academic Press, 2001, pp. 265–300.
8. Brittain HG. Strategy for the prediction and selection of drug substance salt forms. Pharm Tech 2007; 31(10):78–88.
9. Anderson BD, Flora KP. Preparation of water-soluble compounds through salt formation (chap. 34). In: Wermuth CG, ed. The Practice of Medicinal Chemistry. New York: Academic Press, 1996, pp. 739–54.
10. Stahl PH, Wermuth CG. Handbook of Pharmaceutical Salts. Weinheim: Wiley-VCH, 2002.
11. Jacques J, Collet A, Wilen SH. Enantiomers, Racemates, and Resolutions. New York: John Wiley & Sons, 1981, pp. 43–81.
12. Brittain HG. Molecular motion and vibrational spectroscopy (chap. 7). In: Brittain HG, ed. Spectroscopy of Pharmaceutical Solids. New York: Taylor & Francis, 2006, pp. 205–33.
13. Monkhouse DC, Van Campen L. Solid State Reactions—Theoretical and Experimental Aspects. Drug Dev Indust Pharm 1984; 10:1175–276.
14. Byrn SR, Pfeiffer RR, Stowell JG. Solid-State Chemistry of Drugs, 2nd ed. West Lafayette: SSCI Inc., 1999.
15. Carstensen JT. Effect of moisture on the stability of solid dosage forms. Drug Dev Indust Pharm 1988; 14:1927–69.
16. Taylor LS, Zografi G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm Res 1997; 14:1691–98.
17. Al-Nimry SS, Assaf SM, Jalal IM, Najib NM. Adsorption of ketoifen onto some pharmaceutical excipients. Int J Pharm 1997; 149:115–21.
18. Guillory JK, Hwang SC, Lach JL. Interactions between pharmaceutical compounds by thermal methods. J Pharm Sci 1969; 58:301–8.
19. Wirth DD, Baertschi SW, Johnson RA, et al. Maillard reaction of lactose and fluoxetine hydrochloride, a secondary amine. J Pharm Sci 1998; 87:31–9. 20. Forbes RA, McGarvey BM, Smith DR. Measurement of residual isopropyl
alcohol in loracarbef by near-infrared reflectance spectroscopy. Anal Chem 1999; 71:1232–9.
Part 2: Preliminary Preformulation
2.1
Accelerating the Course of
Preliminary Preformulation Through
Prediction of Molecular Physical
Properties and Integrated Analytical
Data Management
Robert S. DeWitte and Michel Hachey
Advanced Chemistry Development, Inc., Toronto, Ontario, Canada
Harry G. Brittain
Center for Pharmaceutical Physics, Milford, New Jersey, U.S.A.
INTRODUCTION
Preformulation research is the necessary step that takes place between drug discovery and clinical development. In this vital phase of drug development, the physicochemical profile of the active pharmaceutical ingredient is determined and then used to plan the course of subsequent activities. One important activity is to plan a schedule of crystallization conditions in order to maximize the likelihood of finding polymorphs or solvatomorphs, since different crystal forms will likely exhibit different solution characteristics that may be either harmful or helpful toward a particular formulation.
Another important activity is to select excipients that enable the drug substance to be administered with its intended efficacy. Preformulation can therefore be described as the step that deals with the acquisition of data on the drug compound and its excipients, which are then analyzed or processed into information (properties and tendencies), and ultimately transformed (modeled) into knowledge in the form of a recommended formulation. The
area of preliminary-preformulation represents the scope of activities that facilitate the design of preformulation studies and is often theoretical in nature.
Although conventional computers cannot match the creativity of the human mind in recognizing, associating, and interpreting data, they can assist scientists by organizing and reducing huge amounts of data to information or knowledge. In the context of preformulation, computers can enhance the consistency and speed of decision making, free experts from tedious tasks, retain the organization’s expertise in a readily maintainable form, and provide knowledge for some specific problem. Computers make desirable outcomes possible through three principal capabilities: (i) effective data management, (ii) information generation and reporting, and (iii) prediction.
Effective data management. Preformulation scientists can benefit from a data management system owing to the wide range and large amount of data generated in their daily work programs. Consider, for example, that the preformulation scientist’s interest in analytical data spans the standard spectroscopic techniques used by organic chemists (such as infrared absorp-tion, Raman spectroscopy, and nuclear magnetic resonance), as well as traditional solid-state characterization techniques (such as X-ray diffraction and thermal analysis). Unless an analytical data management system (ADMS) is used, the late-stage preformulation scientist will not necessarily receive all of the information that has been measured during the conduct of early-stage investigations, or in the associated reports.
Information generation and reporting. A good data management system not only stores data, but also directly analyzes and processes this data into information, which can extend beyond the constraints of a particular technique or chemical property. Information can generally be defined as the relational connection between the data that provides meaning. In other words, it is an answer to a question. Generally, the more connections that can be made, the better equipped one becomes to answer questions. However, the ability to generate intelligent links between chemical struc-tures, properties, and analytical data requires more than a simple relational database. It is highly desirable that the links be active rather than passive. For example, to find all previous reports and database entries related to a particular compound, one may execute a structure search if a real active structure object is used in the data file as opposed to a passive image of the structure. Since reports are the embodiment of answers to questions, flexible publication tools are a necessary facet of an information system for preformulation.
to predict with good accuracy various physical properties (such as log P, pKa, log D, and solubility) from the chemical structure. The computer’s
ability to generate these properties in a short amount of time provides inexpensive, rapid, useful, and early insight into the various preformulation issues that may be encountered. In addition, this early insight allows one to design better experiments at the onset that minimize the amount of work required to satisfy regulatory authorities.
In this chapter, we will introduce two practical methods whereby software can be used to accelerate the pace of preformulation research. The concept of preliminary preformulation is easy to state: to know everything that can be known before beginning a preformulation study. Therefore, we will first review the prediction of the physicochemical properties. Second, we will provide an overview of the advantages of an integrated ADMS and the computational tools that form intelligent links between chemical struc-tures and analytical data to enable an efficient increase of laboratory throughput and information dissemination.
PREDICT BEFORE YOU MEASURE
“Look before you leap into the unknown” is a common maxim that most would agree is good advice. To a large degree, the art and science of mea-suring thermodynamic properties can be accelerated when one has clear expectations. For example, it is possible to minimize the number of buffer solutions needed to explore the entire pH profile for the log D or the solubility of a compound by choosing several pH values over regions of change, and fewer pH values along plateau areas. Some of the key questions, for a given compound, are the following: Where are the ionization centers that give rise to the various pKavalues, and what is the effect of these on the
intrinsic solubility? How many of the predicted ionized forms would be relevant to the formulation of the drug, or to its bioavailability ? Armed with this proto-information, the researcher can select experimental conditions that give precise, useful, and timely measurements when the experiment is performed for the first time.
automatically indicate the molecular site of the ionization, whereas the predictive software would provide that assignment.
Predictive Methods for Molecular Properties
Lipophilicity, pKa, and aqueous solubility, among others, may be regarded
as additive constitutive properties. Such properties are derived from the sum of the intrinsic properties of atoms or functional groups within the molecule, and constitutive properties depend on the structural arrangement of the atoms within the molecule. The full description of these algorithms and their theoretical foundations is beyond the scope of this chapter, and has been published elsewhere (1,2). However, it is appropriate to illustrate the computational approach through a summarization of the approaches used by Advanced Chemistry Development (ACD)/Labs in its predictive software programs.
Calculation of LogP
The octanol–water partition coefficient is widely used as an estimation of the relative lipophilicity of a given compound. The logPalgorithm used by ACD/Labs is based on the well-characterized logPcontributions of separate atoms, structural fragments, and intramolecular interactions between dif-ferent fragments. The basis of the ACD/logPapproach for the prediction of logPcan be described with reference to the following equation:
logP¼Xfnþ X
Fm ð1Þ
wherefdenotes atomic or fragmental increments, andFdenotes correction factors. Figure 1 illustrates the fragments and interactions used in the logP calculation for a tautomeric form of sildenafil.
Using experimental data and a step-regression technique, simple compounds were studied on a class-by-class basis to derive the f and F parameters, and then more complex compounds were studied to derive further fragmental and interaction constants. This process was repeated until the most complex compounds had been used to define the entire listing of fragment increments and interaction patterns. In all cases, fragments were defined by applying the principle of the isolating carbon, a procedure that has been described elsewhere (1). It is important to note, however, that the f and Fparameters so produced can be modeled by fundamental physical organic considerations.
Calculation of pKa
The general structure of the algorithm used in the prediction of ionization constants is one of classification, followed by a linear free energy (Hammett equation) relationship. The method uses sigma constants as descriptors of the electron withdrawal and/or donation potency of substituents electro-nically connected to the ionization center. When more than one ionization center is present in the compound of interest, the researcher must first identify all the separate transitions and their corresponding approximate pKa values in order to discover the correct sequence of ionizations. When
two or more pKavalues are similar in magnitude, multiple scenarios must be
considered in order to understand the perturbing influence of one reaction on another. Doing so will serve to define the observable transitions, as well as the specific ionic form of the acid and conjugate base before and after ionization.
These specific ionic structures define the form of the Hammett equa-tion that should be applied, and/or the appropriate sigma constants to be
N
Figure 1 LogPfragments and interactions for a tautomeric form of sildenafil. (Top) The fragments recognized according to the principle of isolating carbons are shaded.
(Bottom) The aromatic interaction between two of the fragments. In this case the
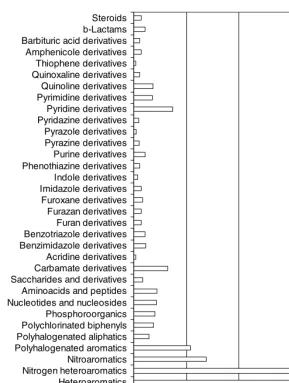
used within the equations. The classes, sigma parameters, and Hammett equations were derived by a study of nearly 16,000 compounds having over 30,000 pKavalues. The composition of this training set is given in Figure 4,
and the correlation between the predicted pKavalues associated with over
12,000 compounds with corresponding entries from the BioByte Masterfile is shown in Figure 5. The latter comparison of predicted pKaparameters for
more than 12,000 experimental values was characterized by anR2parameter of 0.992 and an average error of 0.54.
Aromatic hydrocarbons
Calculation of LogD
The prediction of distribution coefficients (expressed as logD) is performed through an evaluation of the system of multiple equilibria associated with all possible ionic forms that would exist in solution at a specified pH value, and the partitioning of each ionic form between water and octanol. Each ionic form partitions with a different effective log P*. These log P* values are derived from the log P value (which is specifically defined only for the neutral form of the compound) by the addition of specific offset parameters that depend on where the charge is localized on the compound. These offset parameters were derived by studying the log D profiles of multiprotic compounds. As a means of illustration, Table 1 shows the logPcorrection factors corresponding to the ionic forms (i.e., microspecies) existing in the two-phase system that is used for the calculation of logDfor albuterol.
The accuracy of logDpredictions through the ACD/Labs algorithm depends on the accuracy of the pertinent logPcalculation, as well as how close the pH is to one of the pKavalues of the compound. If |pH – pKa|<2,
the accuracy of the pKa term can have a large nonlinear effect on the
accuracy of the logD. If the pH is such that the compound is nearly com-pletely ionized, the accuracy will partially depend on the correction incre-ments such as those in Table 1.
logP(exp) = 0.958 logP(calc) + 0.0969 N = 10895, R2 = 0.9562, AE = 0.213
–6 –4 –2
0
2 4 6 8 10 12 14
–6 –4 2 4 6 8 10 12 14
LogP calculated 7.0
Log
P
e
xper
imental
Calculation of Solubility
The models built into the calculation of solubility are designed to be linear combinations of slowly varying physical functions, with at least an “ansatz” justifying their inclusion (i.e., one can construct a phenomenological defense of the resulting model). Where statistics allow, the models also contain a number of specific fragmental contributions. The resulting form of the models is therefore:
logS¼logPþMWþMVþBPþ"HydBondþFRBonds
þPolarizþjnd20þFrgþTmþconstant ð2Þ
where logSis the intrinsic solubility, mol/L (logarithmic scale); logPis the octanol–water distribution ratio for partially dissociated compounds (loga-rithmic scale); MW is the molecular weight*; MV is the McGowan (3) mole-cular volume*; BP is the boiling point at 760 torr*; HydBond is the hydrogen bond parameter: exp( (NAþND)); NA is the number of H-bond acceptors; ND is the number of H-bond donors (in some tables it is specified as exp ( DA)*); DA is the sum of hydrogen donors and hydrogen acceptors*; FRBonds is the number of free rotatable bonds*; Polariz is the polarizability*; nd20is the refraction index*;SFrg is the sum of fragmental increments; andTm
is the melting point ( ˚C).
The parameters marked by an asterisk were calculated from the chemical structure of the molecule using ACD/PhysChem software. Experimental values of logSand logPwere used to search for the best correlation equations.
0 Other bioactive compounds
Sugars CH acids Other acids Condensed 5- and 6-member heteroaromatics a-amino acids and derivates v 5.0: 8.924 compounds
7000
While models were built using the intrinsic solubilities culled from literature and database sources according to Equation 2, the prediction algorithm was implemented with the augmented formula shown in Equation 3 to allow computation of the pH dependence of the solubility, in which,
pKa(exp) = 0.992 pKa(calc) + 0.08 N = 12126, R2 = 0.943, AE = 0.54 –15
–10 –5 0 5 10 15 20
–15 –10 –5 5 10 15 20
Figure 5 Correlation of the ACD/Labs method for calculation of pKaagainst the
BioByte Starlist of logPmeasurements.
Table 1 LogPCorrection Increments for Different Charge Locations Within the Multiple Ionic Forms Used in Calculating the LogDof Albuterol
Monoanions
Charge on O or S near aromatic –3.15 Charge on O or S near aliphatic –4.10
Monocations
Charge not near aromatic –3.10
Zwitterions
Zwitterionic correction –2.50
Other charged forms
logSðpHÞ ¼logDþMWþMVþBPþ"HydBondþFRBonds þPolarizþjnd20þFrgþTmþconstant ð3Þ
where logDis the octanol–water distribution ratio for partially dissociated compounds (logarithmic scale).
The composition of this training set is given in Figure 6, and results from prediction are given in Figure 7. The comparison with the logSof over 2000 compounds was characterized by anR2parameter of 0.87.
Hydrocarbons
How ACD/Labs Software Presents Predictive Results
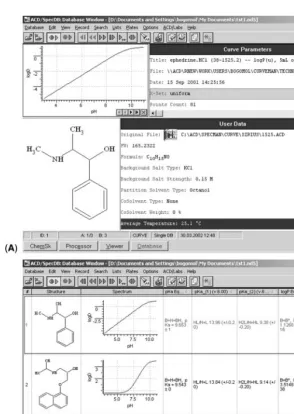
As an example of the computational approach, the use of ACD/Labs predictive software will be employed. Figures 8 through 11 show screen images obtained for the results of calculations performed to predict the molecular physical properties of Indinavir, an antiviral compound. These calculations only required a few minutes on a laptop computer. It is therefore feasible, and highly informative, to calculate the fundamental properties of a new compound and use the predicted values to design experiments before performing any actual lab experiments.
ANALYTICAL DATA MANAGEMENT IN PREFORMULATION
thermal analysis, and at least one solid-state spectroscopic technique. This process ends with identifying the chosen form that will be the subject of continued development.
Once the desired form of the active ingredient is chosen, mixtures of the active ingredient with suitable excipients are subjected to a battery of stress conditions. These stress conditions are designed to reveal possible changes or reactions in the mixtures that indicate the existence of distinct physical or chemical interactions. The stability of these samples is monitored through a variety of techniques including chromatography, optical spec-troscopy, XRPD, and traditional solid-state spectroscopy.