Genetic manipulation of animal cells is extremely important for both basic and applied research. From the perspective of human biology, medical research, and clinical appli- cations, genetic manipulation of human cells and other mammalian cells is especially relevant and important, and so the great bulk of this chapter is focused on this aspect.
But, on occasions, we describe important applications of genetic manipulation of the cells of other animals.
Genetic manipulation of animal cells often involves transferring genetic material into cultured cells, but sometimes the recipient cells are single cells (such as fertilized oocytes) or cells within a living model organism or person. The genetic material is often a genetically engineered DNA construct, and when transferred into cells, the introduced DNA is known as a transgene (even though it may contain multiple genes, or lack any gene). Often, but not always, transgenes are intended to make desired RNA or protein products. Very occasionally, RNA has been transferred into cells (but it is generally pre- ferred to transfer a transgene that is then expressed in the cell to make the desired RNA).
In addition, some applications involve transfer of synthetic oligonucleotides into cells.
The aim may be to study the effect on properties of the transfected cells themselves, or to use the transfected cells as a way of introducing genetic material into an experi- mental organism (transferring genetic material into the germ line to create a transgenic animal), or into a person in an attempt to combat some disease (gene therapy). The moti- vation may be to understand a basic research question—such as: If I inactivate gene X, what are the phenotypic consequences? Or it may be a question of applied research, a commercial application, or a clinical intervention.
In this chapter we describe the basic principles and technologies for transferring genetic material into intact mammalian cells. We will deal with applications of the methodology in other chapters, notably in Chapter 21, when we consider applications in modeling disease, and in Chapter 22 when we consider gene therapy. Table 8.1 gives a brief, and very selective, list to illustrate some of the applications of genetic manipula- tion of mammalian cells.
Transporting naked DNA and oligonucleotides into mammalian cells is normally unnatural, and the cell membrane usually poses a formidable barrier to transport of such large, highly charged macromolecules. However, viruses that infect mammalian cells have refined ways of subverting the usual controls regulating transmembrane trans- port. They are able to efficiently transfer their DNA or RNA genomes into cells, after first surrounding their genomes with a viral protein coat. Viral genomes can be genetically modified—after first converting RNA genomes into complementary DNA, in the case of RNA viruses—and used as vectors to allow the transfer of desired nucleic acid sequences into cells. The alternative is to use nonviral physical or chemical transfer methods. We provide details of the different approaches in Section 8.1.
The introduced genetic material may, or may not, be designed to be incorporated into a cell’s genome. If inserted into a chromosomal DNA, an introduced transgene becomes a stable part of the chromosome, and is transmitted to daughter cells after cell division.
Alternatively, the genetic material may be intended to have an extra chromosomal loca- tion (as is always the case for synthetic oligonucleotides).
According to need, the transfer of genetic material may be intended to produce dif- ferent effects within the intact cell. Transgenes may have been engineered to produce
Principles of genetic manipulation of
mammalian cells 8
functional proteins of interest, or noncoding RNAs. We consider the principles of expressing transgenes in mammalian cells in Section 8.2. Other procedures are designed to make specific, fine-scale changes to a pre-determined target sequence within the genome of intact cells or to specifically suppress expression of a pre-determined gene within the cell.
AN OVERVIEW OF GENOME EDITING, GENE SILENCING, AND GERM-LINE TRANSGENESIS
Genome editing involves making desired changes to the sequence of a pre-determined DNA sequence of interest (the target site) at some specific location within the genome of an intact cell. (Because the target DNA sequence is usually a gene, genome editing has often been described as gene targeting.)
At its most fundamental level, genome editing relies on two components: (1) endo- nucleases to cut both DNA strands at, or close to, a pre-determined target sequence of interest; and (2) protein complexes that rejoin the broken DNA strands. Following genome editing, screening must be carried out to identify those cells where a desired DNA change has been produced. There must, of course, be some way of ensuring a high degree of sequence specificity so that cleavage is directed to occur at a desired target sequence. And genome editing often (but not always) relies on artificial intervention to direct the desired sequence changes.
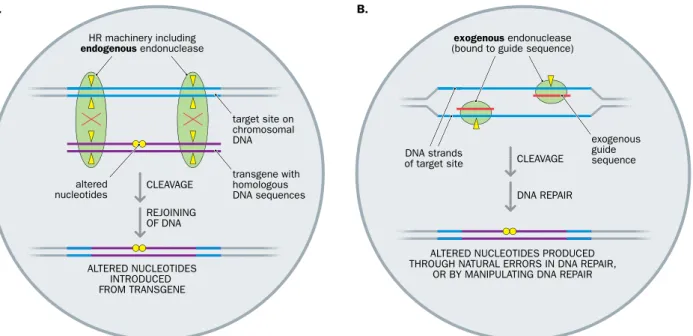
Two quite different genome editing approaches have been used (Figure 8.1).
A traditional method, which has been used in mammalian cells for more than three decades, relies exclusively on homologous recombination using endogenous endonucleases that are part of the cell’s recombination machinery. The method relies on designing the transgene to have long stretches of DNA that show 100%
sequence identity to the target site plus some altered sequence that can be intro- duced into the target site by recombination. The high degree of sequence homology between the transgene and the target site within the genome promotes recombina- tion between them, in which case endogenous endonucleases cleave the DNA at, or close to, the target site (Figure 8.1A). We describe the principle of using standard
TABLE 8.1 SELECTIVE EXAMPLES OF GENETIC MANIPULATION OF MAMMALIAN CELLS
Process Description/comments Gene
expression for protein purification
A transferred human or animal coding DNA is expressed in large quantities in cells that will have appropriate post-translational modification systems.
Mammalian cDNAs can be expressed in well-established mammalian cell lines such as CHO or HEK293 cells. Genetically engineered antibodies are often produced in this way using myeloma cell lines
Directed cell differentiation
Coding DNAs specifying appropriate master transcription factors are expressed in cells of one type causing them to change to a different type. Allows basic studies of cell differentiation and has therapeutic potential (Chapters 21 and 22) Gene transfer to
alleviate genetic deficiency
A coding DNA is expressed to give a product lacking in the recipient cells. Used in some gene therapies where suitable cells of a patient are cultured and then genetically manipulated in vitro before being returned to the patient (Chapter 22) Genome editing
to inactivate a specific gene
Often used to investigate gene function or to make animal models of disease (Chapter 21). The genetic deficiency can be extended to the germ line by manipulating the fertilized oocyte of a model organism or cultured pluripotent stem cells that can be implanted in an embryo to give rise to a model organism with the mutation in every nucleated cell
Genome editing to make altered gene product
May be used to investigate gene function or to model disease and has therapeutic potential
Gene silencing to suppress expression of a specific gene
Relies on transferring oligonucleotides (or antisense RNA) to inhibit expression of a gene that makes an RNA containing the same sequence. A fast and simple way to study the cellular function of a gene, it has also been used to model disease (Chapter 21) and in RNA-based therapeutic approaches (Chapter 22)
homologous recombination in genome editing in Section 8.3. Homologous recom- bination can also be used to direct specific large-scale changes at pre-determined positions in the genome, including very large deletions, large-scale inversions, and translocations (chromosome engineering).
A second, quite recently developed method of genome editing uses programmable site-specific nucleases. Transfected transgenes are expressed to make exogenous endo- nucleases plus protein or RNA guide sequences. The guide sequences are designed to bind to specific sequences at a pre-determined target site within the genome, and are physically bound in some way to the introduced endonuclease. With the exogenous endonucleases attached, the guide sequences bind to their target sequences whereupon the bound endonucleases cut both DNA strands to make a double break (Figure 8.1B).
The resulting double-strand DNA break activates an emergency DNA repair sys- tem (unnatural double-strand breaks in DNA can be fatal for a cell—the priority is to effect repairs rapidly). The method then relies on cellular DNA repair systems to repair the broken DNA. Often an error-prone DNA repair mechanism is used, which by itself introduces a desired change in the target DNA sequence. It is also possible to artificially intervene in the DNA repair to direct desired changes in the target DNA. In either case, treated cells are screened to identify those cells where the altered nucleotides are of the desired type. We describe three popular methods of genome editing using programma- ble endonucleases in Section 8.4.
Genome editing allows all manner of changes to be introduced into target sequences, but it can take some time. Rather than inactivate a gene, it is often more convenient to simply down-regulate the gene. Gene silencing is a fast and simple approach in which RNA transcripts of a pre-determined target gene are targeted to suppress gene expres- sion. In Section 8.5 we describe methods of gene silencing that depend on introducing target RNA-specific oligonucleotides or antisense RNAs into cells.
HR machinery including endogenous endonuclease
transgene with homologous DNA sequences altered
nucleotides
DNA strands of target site
ALTERED NUCLEOTIDES INTRODUCED FROM TRANSGENE
target site on chromosomal
DNA exogenous
guide sequence CLEAVAGE
REJOINING OF DNA A.
exogenous endonuclease (bound to guide sequence)
ALTERED NUCLEOTIDES PRODUCED THROUGH NATURAL ERRORS IN DNA REPAIR,
OR BY MANIPULATING DNA REPAIR CLEAVAGE
DNA REPAIR B.
Figure 8.1 Two very different approaches to genome editing. (A) Genome editing using homologous recombination (HR) alone.
The transgene is designed to have some flanking sequences that are identical to those at a specific target site in the genome but altered sequence (yellow circles) in a central region. Cells are screened to identify rare recombination events between the transgene and target site, notably a double crossover that inserts the altered sequence into the target site (crossover points are indicated by a red X; yellow triangles signify cleavage points). Recombination is carried out by endogenous enzymes, including an endonuclease that cuts double-stranded DNA and a DNA ligase that rejoins the DNA fragments. Details are provided in Section 8.3. (B) Genome editing using programmable exogenous nucleases. Introduced transgenes produce endonucleases plus protein or RNA guide sequences designed to bind to specific DNA sequences at a pre-determined target site in the genome. The exogenous endonuclease is covalently bound to a protein guide sequence or noncovalently to a hybrid RNA that contains a guide sequence plus binding sites for the endonuclease. A pair of sequence-specific guide sequences is used to target bound endonuclease, to cut both DNA strands at a desired unique site. The resulting double-strand break can be repaired rapidly, in which case errors are introduced that may introduce desired sequence changes (yellow circles), or by manipulating an homologous recombination pathway to introduce the desired changes. Different cells may have different altered sequences and are screened for the presence of a desired sequence alteration. Details are described in Section 8.4.
Finally, in Section 8.6, we describe how transgenes can be inserted into the germ line to make transgenic animals. That often involves first transfecting cultured pluripotent cells (embryonic stem cells or induced pluripotent stem cells) or germ-line precursor cells. From the perspective of human genetics, transgenic animals have three particu- larly valuable research applications, as listed below.
• Investigation of how mammalian (and animal) genes function (to allow under- standing of different aspects of cell, tissue, and organ biology).
• Disease modeling. As described in detail in Chapter 21, genetically modified ani- mals are crucially important as models of human diseases, allowing deep insights into the molecular basis of disease.
• As test systems for proposed new disease treatments, especially where genetically modified animals have been shown to be good disease models.
8.1 ARTIFICIAL TRANSFER OF GENETIC MATERIAL INTO MAMMALIAN CELLS
The plasma membranes of our cells are principally organized around hydrophobic lipid bilayers and are semi-permeable: they naturally allow exchange of certain small mol- ecules. In general, the smaller and less polar a molecule is, the higher the chance that it can diffuse across the membrane. Small, nonpolar molecules such as oxygen and carbon dioxide therefore diffuse readily across a lipid bilayer; some small polar molecules, such as water molecules, also diffuse across lipid bilayers, but do so at much reduced rates.
Simple ions, such as Na+, K+, and Ca2+, cannot simply diffuse across the plasma mem- brane. They may be passively transported with the help of concentration gradients or be actively pumped against a concentration gradient. Certain integral membrane proteins known as ion channels help regulate the flow of ions across the cell membrane. Other integral protein-based transporter systems are important in regulating transmembrane transport of some polar molecules such as glucose.
Active transport is needed for macromolecules to cross the plasma membrane and involves a type of endocytosis, the general process in which a portion of the plasma mem- brane invaginates to form a pit and then pinches off to form an endocytic vesicle, enclos- ing some of the extracellular fluid. When endocytosis entraps smaller neighboring cells, such as microbes, the mechanism is known as phagocytosis. Another type of endocytosis, known as receptor-mediated endocytosis and described later in this section, is responsible for internalizing certain proteins and some polysaccharides that bind to a cell surface receptor, and also some viruses that gain entry to the cell by binding cell surface receptors.
The artificial transfer of genetic material into mammalian cells is known as transgenesis. Nonviral or viral methods can be employed to expedite the passage of large, charged nucleic acids and oligonucleotides across plasma membranes. The use of physical or chemical nonviral transfer methods is known as transfection. Viral approaches are modeled on viruses that naturally infect animal cells: here, the nucleic acids are transferred after packaging them into a virus protein coat, and the transfer pro- cess is referred to as transduction.
Note that the terminology differs from that used in bacterial systems, where transfection means the uptake of naked virus DNA, while the uptake of naked plasmid or genomic DNA is described as transformation. (But for mammalian cells, transformation is a term reserved for changes in genotype, with ensuing changes in cell attributes and behavior associated with cancer.)
Depending on the delivery system used to get them into cells, transgenes may be designed to migrate to the nucleus and, in some cases, insert into a chromosomal DNA.
Otherwise, a cytoplasmic location may be sufficient for their function. As described below, a cytoplasmic function is often intended also for RNA and oligonucleotides trans- ferred into cells. The cells that are transfected are often cultured cells. As we describe below, different procedures have been developed to make stable permanent cell lines that can be disseminated to laboratories throughout the world. Before examining meth- ods of transfecting genetic material into mammalian cells, we first look at how mam- malian cells are cultured.
Culturing mammalian cells: primary cell cultures, cell culture methods, and cell storage
Establishing cultured cells begins by surgically removing cells from an organism and placing them into a suitable culture environment. For that, a suitable vessel is needed, made of glass or treated plastic, and a culture medium containing nutrients to help
cells grow. The medium contains a buffer to maintain an appropriate pH (often pH 7.0–7.4) and culture vessels are maintained at a suitable constant temperature (usually 37°C), often within an incubator designed to provide an atmosphere of 5% CO2. After a short period, cells will attach to the surface or a substrate layer on the surface of the cul- ture vessel, then divide and grow to form a primary culture. There are two primary routes toward achieving this objective, as listed below.
• Explant cultures. Small pieces of tissue are attached to a glass or plastic vessel and bathed in culture medium. After a few days, individual cells will move from the tissue explant onto the culture-vessel surface where they begin to divide and grow.
• Enzymatic dissociation. In this widely used method, proteolytic enzymes (trypsin, collagenase) are first used to dissociate tissue fragments. The ensuing suspension of single cells is placed into culture vessels containing culture medium, and cells are allowed to grow and divide.
After cells in the primary culture vessel have grown to fill up all of the available cul- ture substrate, the cells must be subcultured to give them room for continued growth.
Cells are gently removed from the substrate using proteolytic enzymes (to break protein bonds attaching the cells to the substrate), and the suspension of recovered cells is then subdivided and placed into new culture vessels.
Two basic cell culturing methods are employed, according to whether or not cells are able to adhere to a glass or treated plastic substrate. If they are able to do so, monolayer cultures are established, and different vessels are possible: tissue culture treated dishes, T-flasks, roller bottles, multiple-well plates, and so on. If, instead, the cells float freely, they are often kept actively suspended in the medium (suspension cultures—the cells are grown in magnetically rotated spinner flasks or in shaken flasks).
Cells may be maintained by passaging (or splitting): a small number of cells from a previously grown culture are seeded into fresh culture medium in a new culture vessel.
If cells are passaged regularly, cell senescence (associated with high cell density) can be avoided and the cells can be cultured for longer.
Cell line storage and testing
Cell lines may be stored in a frozen state after adding some kind of cryopreservative agent—often DMSO (dimethylsulfoxide)—to the cell medium. As described in the fol- lowing section, the most useful cell lines are immortalized cell lines that can be cultured indefinitely (they can be temporarily stored frozen in liquid nitrogen; thawing a frozen sample yields a viable culture). Certain major centers serve as cell line repositories—see Table 8.2 for some examples. There is a need to periodically check the authenticity of cell lines (by DNA fingerprinting) because proximity of other cell cultures in laboratories often leads to overgrowth of cultured cell lines by other, faster-growing cell lines.
TABLE 8.2 EXAMPLES OF MAJOR CELL LINE REPOSITORIES
Cell line repository Website
ATCC—American Type Culture Collection http://www.lgcstandards-atcc.org/
Coriell Institute Biorepositories https://catalog.coriell.org/
ECACC—European Collection of Authenticated Cell Cultures
https://www.phe-culturecollections.org.uk/
collections/ecacc.aspx
The utility and construction of immortalized cell lines
Early cell lines had a limited lifetime because cells develop senescence after a finite number of cell divisions (the Hayflick limit, as described in Section 3.2). Immortalized (permanent) cell lines have the huge advantage of allowing cells to be cultured indefi- nitely. As well as allowing applications that last over long timescales, and avoiding hav- ing to devote significant effort into just keeping the cells alive, there is the convenience of being able to access the desired cells from major cell line repositories.
Cells can become immortal when mechanisms regulating cell division are subverted, allowing cells to keep on dividing. That happens naturally in cancers:
normal cell division controls are dysregulated following mutations in certain cancer- susceptibility genes, or after transformation of cells by cancer-causing viruses.
Some popular permanent cell lines were originally obtained from naturally occur- ring tumors (such as the HeLa human cell line that originated from Henrietta Lacks, a patient with cervical cancer). And after being identified as key components in cellular transformation, oncogenes could be artificially expressed in cultured cells to induce transformation, resulting in immortalized cell lines. For example, many immortalized cell lines have been made by transfecting and expressing an oncogene from simian virus 40 (SV40). The gene product, the SV40 large T antigen, binds to and inhibits p53 and the pRb retinoblastoma protein, proteins that normally act as brakes on cell division.
Immortalized cell lines derived from tumors or artificially transformed cells are dis- advantaged by genome instability, and aneuploidy is common. Molecular pathways in transformed cell lines, therefore, may not always be representative of those in the origi- nal untransformed cells. But for some purposes that is not a major concern. The major utility of immortal human lymphoblastoid cell lines, for example, is to provide an inex- haustible source of DNA from individuals of interest (Box 8.1).
Most people across the world become infected at some stage in their lives with Epstein–Barr virus (EBV—a gamma herpesvirus) and show no symptoms (after gaining adaptive immunity), but the virus occasionally causes glandular fever. EBV can also transform cells that it infects: it is implicated in different cancers, including certain lymphomas, and it also readily infects resting B lymphocytes in vitro, causing growth transformation.
A sample of peripheral blood is sufficient, therefore, to prepare transformed B cells. As EBV becomes integrated into B cells, cytotoxic T lymphocytes can be generated that would kill the infected B cells leading to transforma- tion failure. Accordingly, either the T cells are suppressed, or they are removed in some way before the B cells are infected by EBV (Figure 1).
Following transformation, a lymphoblastoid cell line (LCL) is produced. Initially, the EBV-transformed cells are in a preimmortal stage: the cells are actively prolif- erating and euploid, with no tumorigenic properties.
The telomeres shorten with each round of cell division, but the transformed cells do not undergo senescence at the stage when untransformed B cells would be expected to do so. Nevertheless, after several further rounds of cell division, the cells undergo a stage known as proliferative crisis, during which the vast majority of the cells die. The very rare survivors are cells that have undergone a series of very significant genetic and epigenetic changes, including development of aneuploidy and activation of telomerase.
Because these aneuploid cells can proliferate indefinitely, they are described as immortalized LCLs.
LCLs have been used in a variety of functional assays, but as well as providing a continuous in vitro source of cells, immortalized LCLs have routinely been used to provide an inexhaustible supply of genomic DNA from individuals of interest. Immortalized LCLs have been developed from large numbers of individuals with genetic disorders and within genetic epidemiology studies, allowing detailed genotype–
phenotype correlation studies.
BOX 8.1 MAKING EBV-TRANSFORMED LYMPHOBLASTOID CELL LINES
Box 8.1 Figure 1 Constructing lymphoblastoid cell lines (LCLs) by EBV transformation of B lymphocytes. Traditional construction of LCLs has involved separation of lymphocytes by density
centrifugation of whole blood. Thereafter, some method is used to remove or suppress T cells. Often an immunosuppressant such as cyclosporin is added to prevent T-cell proliferation, or paramagnetic beads coated with an antibody specific for B cells are used to bind B cells and enrich them using a magnetic sorter. Following mixing with Epstein–Barr virus (EBV) particles (usually maintained and isolated from marmoset cells), B cells may be infected by EBV. Transformed cells are cultured and are initially euploid, but if they go through a sufficient number of population doublings, they go through proliferative crisis after which survivors are aneuploid cells that express telomerase and can proliferate indefinitely (immortalized LCLs). Note that more recently developed methods allow
immortalized LCLs to be conveniently isolated from small volumes of cryo-preserved blood (see Amoli MM et al. [2008]; PMID 18381392).
(Adapted from Sie L et al. [2009] J Neurosci Res 87:1953–1959; PMID 19224581. With permission from Wiley-Liss, Inc., © Wiley-Liss, Inc.) B & T
lymphocytes
B-cell separation or T-cell immuno- suppression
transformation
longer-term culture short-term
culture and storage
B B B T
T T
B T
plasma Ficoll larger blood cells
density centrifugation of whole blood in
Ficoll gradient
pre-immortal euploid LCL
CRISIS telomerase activation IMMORTALIZED
LCL
particlesEBV
B B B T B
B B B
T T
B T
Immortalized euploid cell lines
To make immortalized euploid cell lines that are more representative of the original cells, interest has focused on a final step in cell transformation: activation of TERT, telomerase reverse transcriptase. Recall from Section 2.4 that the DNA of telomeres consists of tandem repeats of the hexanucleotide TTAGGG, and that telomerase extends telomeric DNA by using its RNA component, TERC, to provide an RNA template for the TERT enzyme to make new TTAGGG repeats (see Figure 2.25). Only a very few of our cells normally express telomerase because although the TERC RNA is expressed in all cells, the TERT enzyme is normally restricted to unspecialized cells in early develop- ment and to immortal stem cells that are needed to replenish body cells. Normal body cells can undergo just a limited number of cell divisions because DNA is lost at the telo- meres at each round of DNA synthesis (the chromosome end-replication problem; see Figure 2.24), causing progressive shortening of telomeres and eventually inducing cell senescence.
Transformed cells do not undergo senescence. But after several rounds of cell division, past the stage where senescence develops in the normal cells, trans- formed cells undergo a proliferation crisis: the very few surviving cells are aneu- ploid cells that have reactivated telomerase to become immortal. The long route that transformed cells take to achieve immortality can be bypassed by just recreating the last step: by artificially expressing a TERT transgene in cultured euploid cells, it is possible to create euploid cell lines with telomerase activity that are effectively immortal.
Nonviral methods of transferring genetic material into mammalian cells
Nonviral methods are used to transfer different types of genetic material into cells. They are quite inefficient by comparison with viral methods, but because they are often quite simple and convenient methods, they have been widely used when the efficiency of transfer is not the highest priority. In the case of gene therapy, concerns about the safety of using virus vectors to transfer transgenes into the cells of a patient have also prompted interest in the alternative of nonviral transfer methods.
Physical methods
Physical methods are used to transfer genetic material into human or animal cells, either by piercing the cell membrane in some way, or by inducing pores in the mem- brane that allow passage of the nucleic acids or oligonucleotides. Some methods have specialized uses. For example, microinjection of DNA, using a very fine needle to pierce the cell membrane, is limited to transfecting single cells at a time. A common application is to transfer DNA into fertilized oocytes (an important way of delivering genes into the germ line to make transgenic animals). Some of the more generally applicable physical transfer methods are described briefly below.
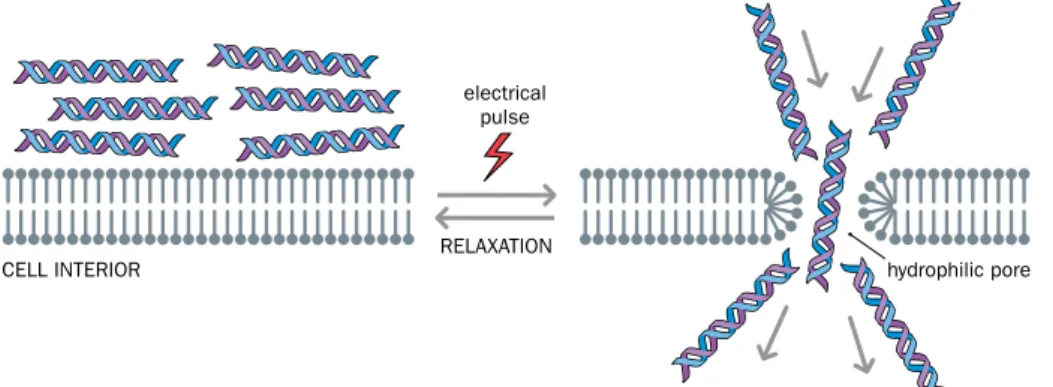
• Electroporation. When exposed to a sufficiently strong electric field, the plasma membrane of a cell undergoes electrical breakdown. Pores form, allowing pas- sage of molecules that normally cannot cross the membrane (Figure 8.2). If the exposure is sufficiently short, the membrane can rapidly recover and become semi-permeable again. Electroporation (short for “ electric pore formation”)
RELAXATION electrical
pulse
CELL INTERIOR hydrophilic pore
Figure 8.2 Electroporation as a way of permeabilizing the plasma membrane.
Although the plasma membrane is a highly fluid structure, it is normally a formidable barrier to nucleic acids and oligonucleotides (which are negatively charged macromolecules). Electroporation involves exposing the cell membrane to very brief pulses of a high voltage electric field, causing pores to form transiently.
Hydrophilic pores are bounded by the phosphate groups of membrane phospholipids and facilitate entry of nucleic acids or oligonucleotides into the cell.
involves administering extremely brief pulses of very high voltage to the mem- branes, allowing entry of desired large molecules and then resealing of the membrane.
• Sonoporation. An alternative way of transiently making pores in membranes to increase permeability is to use very brief pulses of high-energy ultrasonic sound.
• Particle bombardment. Biolistic methods use a gene gun to fire high-density (gold or tungsten) microparticles coated with nucleic acid; the microparticles are accelerated to very high velocity, usually using compressed gas, allowing efficient transfection of cells, irrespective of cell type. Developed initially to transform plant cells (whose cell walls pose a formidable barrier to passage of macromol- ecules), particle bombardment has been used to transfect plasmid recombinant DNA into a variety of cultured mammalian and animal cells, as well as tissues in vivo. The method is especially useful for transfecting cells that are more resis- tant to transfection by other methods.
Chemical methods
Certain chemicals can facilitate uptake of genetic material into mammalian cells by endocytosis. Calcium phosphate has long been used as an aid to gene transfer into mammalian cells. It relies on formation of DNA–calcium phosphate co-precipitates that are adsorbed onto the surface of target cells at high concentration, facilitating uptake by the cells through endocytosis. The transfection efficiency is not very high, however.
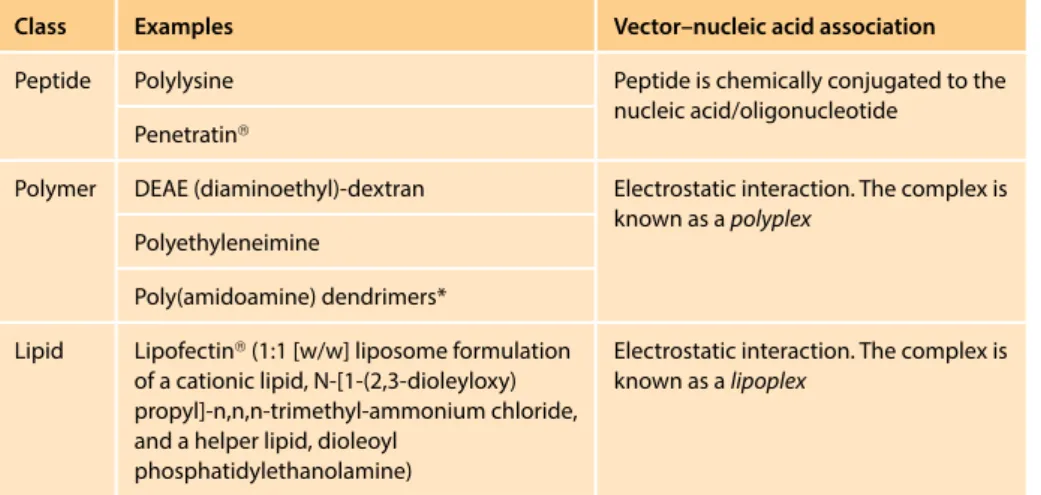
Various other chemical methods rely on cationic (positively charged) macromol- ecules that bind the negatively-charged nucleic acid or oligonucleotide molecules and target them to the cell membrane to facilitate their uptake into cells by endo- cytosis. The vector–nucleic acid complexes have positively-charged surfaces and are attracted to cell membranes whose outer surfaces have numerous negatively- charged phosphate and sulfate groups (within membrane-bound glycoproteins and membrane phospholipids).
Of the wide range of cationic vectors (Table 8.3), cationic lipids, as part of arti- ficial lipid bilayers, have been especially widely used because of their compara- tively high efficiency. This type of transfer (lipofection) uses synthetic spherical vesicles, known as liposomes, that have at least one lipid bilayer and form spontane- ously when certain lipids are mixed in aqueous solution. After the desired nucleic acids or oligonucleotides are combined with a mixture of a cationic lipid and a helper lipid in water, cationic liposomes spontaneously form with bound nucleic acid/ oligonucleotide. After association with the cell membrane, they can be taken up into the cell by endocytosis (Figure 8.3). The efficiency may be increased still further when some other chemical vectors, such as polylysine, are also included in the mix.
TABLE 8.3 EXAMPLES OF POSITIVELY-CHARGED CHEMICAL VECTORS FOR TRANSFERRING GENETIC MATERIAL INTO ANIMAL CELLS
Class Examples Vector–nucleic acid association
Peptide Polylysine Peptide is chemically conjugated to the
nucleic acid/oligonucleotide Penetratin®
Polymer DEAE (diaminoethyl)-dextran Electrostatic interaction. The complex is known as a polyplex
Polyethyleneimine
Poly(amidoamine) dendrimers*
Lipid Lipofectin® (1:1 [w/w] liposome formulation of a cationic lipid, N-[1-(2,3-dioleyloxy) propyl]-n,n,n-trimethyl-ammonium chloride, and a helper lipid, dioleoyl
phosphatidylethanolamine)
Electrostatic interaction. The complex is known as a lipoplex
* So called because they are highly branched structures.
A.
B.
recycling endosome coated vesicle coated
pit
coat protein
ligand
multivesicular body
intralumenal vesicle FUSION
FUSION
MICROTUBULE-MEDIATED TRANSPORT
lysosome
endolysosome late
endosome early
endosome pit vesicle
Golgi network plasma membrane
CYTOSOL
CYTOSOL receptor
Figure 8.3 Receptor-mediated endocytosis and endosome maturation. (A) Receptor-mediated endocytosis. Different classes of protein on the cell surface act as receptors for specific ligands including signaling proteins and viruses. Invagination of the plasma membrane after ligands have bound to receptor proteins leads to formation of pits initially, and then small vesicles that transport the entrapped proteins or viruses within the cell. The pits are coated with a protein, such as clathrin, that plays a major role in vesicle formation, and forms a polyhedral lattice surrounding vesicles. Blue arrows indicate the direction of constriction as the plasma membrane invaginates.
(B) General scheme for endosome maturation. Endocytic vesicles fuse with an early endosome, a sorting station composed of membrane- limited tubules and vesicles. Some membrane proteins, such as receptor proteins, can be returned back to the plasma membrane for reuse, or be stored temporarily in recycling endosomes before being returned. Other membrane proteins and phagocytosed cells are transported via a multivesicular body to a late endosome; further sorting can occur that can lead to fusion with lysosomes and destruction of their contents. (A, adapted from Campbell NA & Reese JB [2008] Biology 8th edn. Pearson/Benjamin Cummings; B, from Alberts B et al.
[2014] Molecular Biology of the Cell, 6th edn. Garland Science. With permission from WW Norton.)
Intracytoplasmic passage and nuclear entry
After passage through the cell membrane, transfected nucleic acids/oligonucleotides may need to escape from endosomes (if taken up by endocytosis). When taken up by endosomes, complexes containing bound nucleic acids or oligonucleotides might be expected to be quickly degraded: the endosome containing them would be shunted into the pathway that leads progressively toward formation of a late endosome and fusion with a lysosome (see Figure 8.3B). That fate can be avoided by designing the com- plex to destabilize the endosome, allowing the transfected genetic material to escape.
Figure 8.4 shows how that is achieved in the case of lipofection.
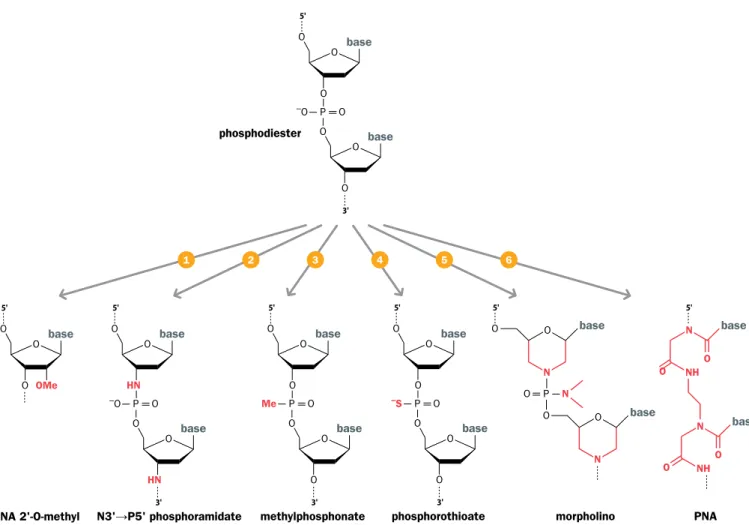
After escaping from the endosome, the genetic material is released from its protec- tive chemical coat and becomes vulnerable to degradation by cytoplasmic nucleases (a defense system against invading viruses). To minimize degradation, oligonucleotides
that are intended to work in the cytoplasm (to block RNA expression) are designed to be robust, nonstandard, synthetic oligonucleotides that are resistant to nucleases (Figure 8.5). For transgenes, the nucleus offers a safer environment, but whereas some virus vectors readily gain access to the nucleus, and even integrate into chromosomal DNA, using nonviral transfer to get a transgene into the nucleus is less straightforward (transport through the nuclear pores is generally inefficient).
To facilitate nuclear targeting, various nuclear localization signal (NLS) peptides were developed to assist active transport of transgenes through nuclear pore complexes.
Subsequently, however, nucleofection, a proprietary modification of the electroporation method using cell-type-specific reagents, was developed by the Amaxa company and has been used with considerable success to transfect transgenes into both the nucleus and cytoplasm. This method has been particularly useful for transfecting a wide variety of nondividing cell types, including neurons.
– + + + +
+ +
+ +
+ + + + + – – –
–
– – – – –
– – – –
– – – –
– – – – – – – – –
– – –
– –
– – – – – –
– –
– – – – – – +
+ +
++ + + +
+ + + + + + +
+ + +
+ + + ++ + + + + + – –
– –
– – – –
– –
lipoplex BINDING
NONCOATED PITMEDIATED ENDOCYTOSIS
endosome
COATED PITMEDIATED ENDOCYTOSIS
COMPLEX DESTABILIZATION
CYTOPLASMIC DELIVERY NUCLEAR
TRAFFIC CYTOSOL
NUCLEUS
neutral lipids anionic lipids cationic lipids
dioleoylphosphatidylethanolamine DNA
Figure 8.4 Cationic liposomes as vectors for delivery of nucleic acids into mammalian cells. The nucleic acid to be transferred is complexed with liposomes to form lipoplexes that have positive charges on the surface, helping interaction with cell membranes (which have multiple negative charges on the surface). The lipoplexes are taken up by cells through different endocytosis pathways in which the cell membrane invaginates to form a pit. Large lipoplexes are taken up by pits coated with clathrin complexes (coated pit-mediated endocytosis at top right); small lipoplexes are taken up by noncoated pits (top left). In either case, the lipoplexes become trapped in endosomes (simplified here; see Figure 8.3B for a fuller picture) and would be expected to be targeted for destruction by lysosomes. However, the inclusion within the liposomes of certain helper lipids—usually electrically neutral lipids, such as dioleoyl phosphatidylethanolamine—helps to destabilize the endosomal membranes, causing the passenger nucleic acid to escape to the cytoplasm (yellow arrows). For a transferred DNA to be transcribed, it must pass to the nucleus. In dividing cells, the breakdown of the nuclear envelope during mitosis allows the DNA to gain access to the nucleus, but in nondividing cells the precise mechanism of entry into the nucleus is unclear. (From Simões S et al. [2005]
Expert Opin Drug Deliv 2:237–254; PMID 16296751. With permission from Informa Healthcare.)
Transgene size range
As general methods for delivering genetic material to mammalian cells, nonviral transfer methods have two principal advantages over viral methods. First, they can readily transfer any type of genetic material—including short RNA molecules or chemically-modified oligonucleotides (viral methods are especially used to trans- fer DNA, but viruses may convert DNA to RNA for propagation purposes). Secondly, nonviral methods can be used to transfer extremely large molecules: it has been pos- sible to introduce transgenes containing megabases of DNA into human cells, where they replicate independently and behave as artificial chromosomes (an example is described in Chapter 21).
Viral methods of transferring DNA into mammalian cells
Over long periods of evolution, viruses have refined ways of packing their genomes into protective protein coats and injecting them into cells. According to the virus, the genome can be DNA or RNA and either single-stranded or double-stranded (Box 1.2 describes the extraordinary variety of viral genomes). Viruses are most readily manipulated as double-stranded DNA molecules to which a DNA of interest can be covalently attached, forming a transgene that can be packaged into a viral protein coat and transported into cells. In the case of RNA viruses, double-stranded DNA copies of the RNA genome (which naturally exist during the viral life cycle as replicative form DNA) are used. If required, the transferred DNA can be a copy of an RNA of interest (artificially made using a reverse transcriptase).
Viral transfer methods offer multiple advantages. First, they allow much higher trans- fer efficiency than nonviral methods. Secondly, according to the type of virus, transgenes
O
O O –O P
O base
O
O O base
5'
3'
O
O O –S P
O base
O
O O base
5'
3' O
O O P Me
O base
O
O O base
5'
3' O
HN O –O P
O base
O
HN O base
5'
3'
O
base
5'
NH N
O O
base
NH N
1 2 3 4 5 6
O O OMe
O
O base
5'
base
N O P O
O 5'
N O
base
N O phosphodiester
phosphorothioate methylphosphonate
N3'→P5' phosphoramidate morpholino PNA
RNA 2'-O-methyl
Figure 8.5 Chemical modification can increase oligonucleotide stability. The standard oligodeoxyribonucleotide structure is shown at the top. Four of the six modified oligonucleotides have a minor change, involving either replacement of atoms directly linked to carbons 2′ or 3′ of the deoxyribose (1,2) or replacement of an oxygen ion of the connecting phosphate (3,4). The other two modifications (5,6) are radical alterations to the normal structure, producing morpholino oligonucleotides or peptide nucleic acids (PNA). (Modified from Dias N & Stein CA [2002] Mol Cancer Ther 1:347–355; PMID 12489851.)
can be ferried into the cytoplasm or nucleus; in the latter case, retroviruses (RNA viruses that replicate through a DNA intermediate) allow integration of a transgene into the genome (retroviral integration into a host-cell chromosome is mandatory for success- ful completion of the life cycle). As a result, a transgene can shelter in the stable envi- ronment of chromosomal DNA and be inherited when cells divide. Additionally, certain strains of viruses are also suited to infecting particular types of cell.
Viral transfer methods do have some downsides. Although their protein coats protect the transferred DNA from nuclease attack, they impose size limits on the DNA that can be transferred—sometimes the maximum limit is just a few kilobases of DNA. And in the case of gene transfer in vivo there can be safety concerns. We will explore these in detail when we consider gene therapy in Chapter 22.
Transduction using retroviral vectors
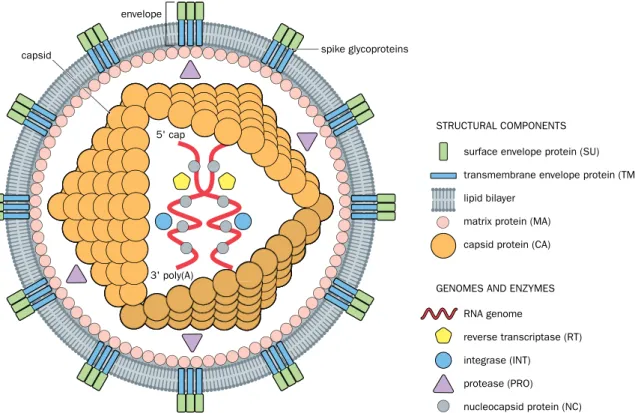
A retrovirus has a single-stranded RNA genome but replicates in the host cell through the process of reverse transcription (in which the RNA is converted to DNA). A com- plete retrovirus particle (virion) has two copies of the RNA genome enclosed within a capsid protein coat; in turn, the capsid is surrounded by a lipid bilayer envelope with spike glycoproteins on the outside. In addition to different types of structural protein, the virion contains three key enzymes: a reverse transcriptase, a protease, and an integrase (Figure 8.6).
3' poly(A) 5' cap
surface envelope protein (SU) transmembrane envelope protein (TM) lipid bilayer
matrix protein (MA) capsid protein (CA)
capsid spike glycoproteins
envelope
RNA genome
reverse transcriptase (RT) integrase (INT)
protease (PRO) nucleocapsid protein (NC) STRUCTURAL COMPONENTS
GENOMES AND ENZYMES
Figure 8.6 Retrovirus structure. An infectious retrovirus particle (virion) has two copies of a single-stranded RNA genome, each with an m7GpppG cap at its 5′ end and a 3′ poly(A) tail (the genome is said to be a positive single-stranded RNA because in the cytoplasm the same RNA serves as a sense strand for making proteins). The genomic RNA is bound and structured by a nucleocapsid protein, and is enclosed within an inner capsid along with some viral enzymes (described in Figures 8.7 and 8.8). The capsid and its contents constitute the core particle, which is surrounded by an envelope consisting of a lipid bilayer with attached proteins. In addition to structural matrix proteins, the envelope periodically has spike glycoproteins consisting of an outer surface glycoprotein (which binds to specific receptors on the surface of cells) and a transmembrane glycoprotein (which aids virus entry into a cell by triggering fusion between the virus lipid bilayer and the cell’s plasma membrane).
An infectious retrovirus particle enters a cell after its surface envelope glycoproteins bind to specific receptors on the cell surface (such as the CD4 receptor in the case of human immunodeficiency virus, HIV). Usually the transmembrane envelope glycopro- tein then triggers the fusion of the viral and cellular membranes, allowing the virus to enter as a core particle, lacking the outer envelope; see Figures 8.6 and 8.7. (But some ret- roviruses enter by the receptor endocytosis mechanism described in Figure 8.3.) After infecting a cell, the core virus uses its multifunctional DNA polymerase to make a com- plementary DNA copy of its RNA, degrade the original RNA, and then copy the surviving single-stranded DNA (see Figure 8.7).
core virus particle infecting virus (virion)
viral RNA provirus cellDNA
NUCLEUS
CYTOPLASM 1
2
4
5
7
8
9 6
LTR LTR
R
R ssRNA
dsDNA 3a
3b 3c
LTR LTR
Figure 8.7 Retroviral life cycle. Infection begins when the virus envelope surface glycoprotein recognizes specific receptors located on the cell surface (1) and the virus enters the cell, usually as a core virus particle that lacks the outer envelope. Thereafter, the viral RNA is released from the capsid (2) and the viral DNA polymerase converts the single- stranded (ss) RNA genome into a double-stranded (ds) DNA (3). That is possible because the viral DNA polymerase is multifunctional, having: a reverse transcriptase activity (uses the ssRNA to synthesize a complementary DNA—step 3a); an RNase H activity (degrades the RNA—step 3b); and a standard DNA polymerase activity (converts the ssDNA to a dsDNA—step 3c). After entry into the nucleus, the viral DNA inserts into chromosomal DNA using the viral integrase enzyme (4), enabling the viral genome (now called the provirus) to be stably maintained, replicated during DNA synthesis, and passed to progeny cells. Viral RNA is produced (5) and migrates to the cytoplasm (6) where it is translated to produce viral structural proteins and enzymes (7). Viral RNA also associates with some newly-produced viral proteins to form new core particles (8). The core particles then obtain their envelopes and are released from the cell (budding; step 9). Mature progeny virions are then capable of infecting new cells. LTR, long terminal repeat; R, short repeat in LTR (see Figure 8.8B).
The resulting double-stranded DNA can be incorporated into the host-cell genome using the viral integrase. The integrated virus, known as a provirus, may remain in the host-cell genome and be transmitted to daughter cells following cell division. If germ- line cells are infected, the virus may be transmitted vertically, but horizontal trans- mission is the norm: after a virus infects a cell, the transcriptional and translational machinery of the host cell is hijacked to produce new copies of the viral genome plus viral proteins, after which new virus particles are assembled and then exit from the cell to infect other cells (see Figure 8.7).
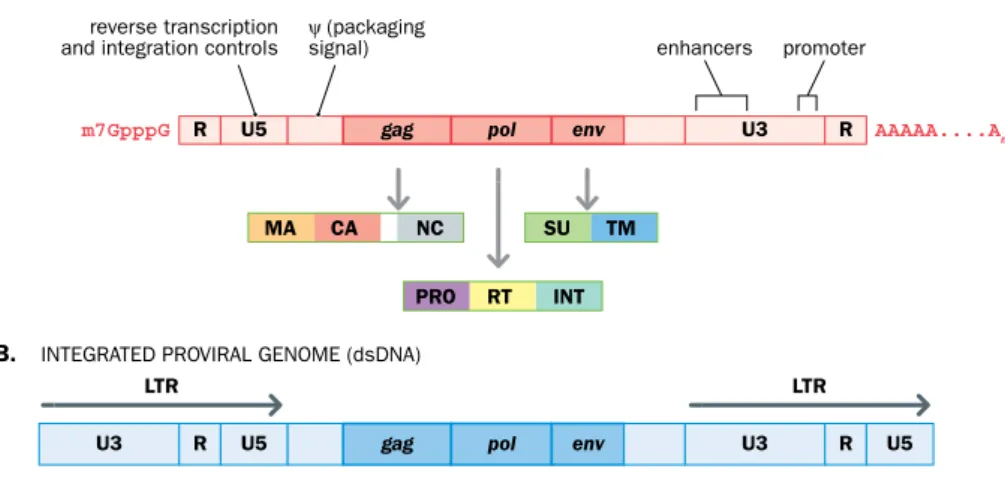
Retroviruses have small genomes, from 7 to 10 kb in length. Simple retroviruses, such as gammaretroviruses, have three genes: gag, pol, and env. They each make protein pre- cursors (polyproteins) that are cleaved to produce two or three individual proteins. In addition, various regulatory sequences are found in the flanking sequences (Figure 8.8).
More complex retroviruses, including lentiviruses such as HIV, have extra genes involved in replication in addition to the gag, pol, and env genes.
enhancers ψ (packaging
signal) reverse transcription
and integration controls promoter
gag U3
LTR LTR
U3
R U5 pol env R U5
gag U3
R U5
MA CA NC
PRO RT INT SU TM
R AAAAA....An
m7GpppG pol env
A.
B. INTEGRATED PROVIRAL GENOME (dsDNA) RETROVIRAL GENOME (ssRNA)
Figure 8.8 Functional components of a simple retroviral genome. (A) The positive single-stranded RNA genome in a virion has a 5′ cap and a 3′ poly(A) tail because the same RNA strand is also translated in the cytoplasm to make viral proteins. Each of the three genes makes a polyprotein precursor that is cleaved to give the individual proteins (see Figure 8.6 for the protein names and locations in the virus). The gag (group antigen) gene makes various structural proteins. The pol gene gets its name because one of its products is a multifunctional DNA polymerase: it can use both RNA templates—a reverse transcriptase (RT) activity—and also DNA templates. The pol gene also encodes a protease (PRO) and an integrase (INT), the enzyme used to insert the viral genome (as double-stranded DNA) into the host cell’s nuclear genome. The env gene makes the two envelope proteins present in the spike glycoproteins (see Figure 8.6). The retroviral protease cleaves the polyproteins encoded by gag and pol;
cellular proteases are responsible for cleaving the env-encoded polyprotein. The flanking sequences contain a short repeat (R) plus some regulatory sequences, including U5 and U3 sequences (in the 5′ and 3′ untranslated regions, respectively) and psi (ψ), a crucial packaging signal that directs incorporation of the RNA genome into virions, which is located downstream of U5. (B) When the RNA genome is converted to double-stranded DNA, the R repeat is responsible for transfer of DNA synthesis between templates so as to cause duplication of the U5 and U3 sequences. As a result, the proviral genome has long terminal repeats, each containing a U5 and U3 sequence as well as the R sequence.
Different classes of retroviral vector are used to transfer transgenes into cultured mam- malian cells, but vectors based on gammaretroviruses, such as murine leukemia viruses, have been widely used. They cannot pass through nuclear pores, and so cannot be used with nondividing cells, but are able to access the nucleus of dividing cells after the nuclear membrane dissolves in preparation for mitosis and then re-forms. Initially, gammaretro- virus vectors were commonly used in gene therapy, but safety concerns associated with their use have prompted the alternative use of lentivirus vectors for this application.
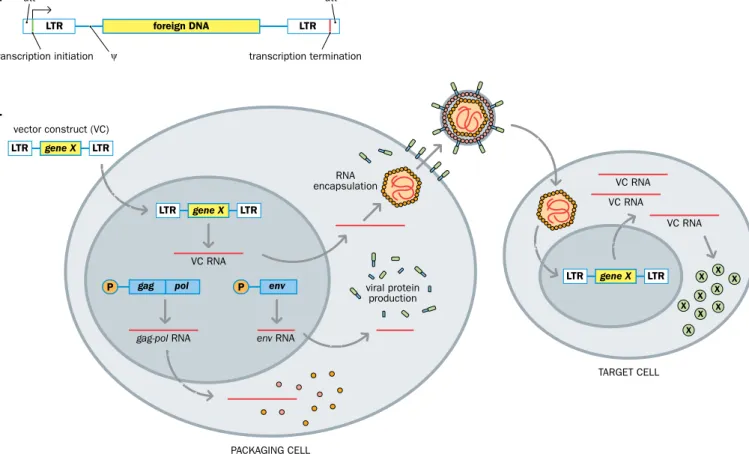
Standard recombinant gammaretrovirus vectors are made by using genetic engineer- ing to modify a double-stranded gammaretroviral DNA, replacing retroviral sequences to create replication-defective vectors. The early steps in the retroviral life cycle—retroviral entry into cells, reverse transcription of the viral genome into DNA, and integration of the viral genome into the host genome—do not require viral protein. As a result, all viral cod- ing regions can be deleted (and replaced by a desired transgene) and the remaining viral sequences can be reduced to the minimum required for high-efficiency transfer. The viral proteins do need to be supplied in trans and a packaging cell line is required to build a virus coat for a vector containing the desired foreign DNA. Virus particles obtained from such cells can then be used to introduce the desired DNA into a target cell of choice (Figure 8.9).
ψ
LTR LTR
vector construct (VC)
transcription initiation transcription termination att att
viral protein production encapsulationRNA
PACKAGING CELL
TARGET CELL VC RNA
VC RNA VC RNA
VC RNA
gag-pol RNA env RNA
gene X
LTR foreign DNA LTR
LTR gene X LTR
LTR LTR X X
X X X X X X
X gene X
gag pol
P P env
B.
A.
Figure 8.9 Retroviral vectors are produced after first transfecting the recombinant retroviral DNA into a packaging cell line. (A) A transgene that is a recombinant retrovirus, often called a vector construct (VC). The gag, pol, and env genes have been deleted and replaced by a foreign DNA of interest, but the viral regulatory sequences have been retained. They include promoter/enhancer sequences, transcription termination signals, and the att (integration) and ψ (packaging) signals. (B) Packaging cell lines are prepared by transfecting a suitable cell with viral genes that can supply suitable viral proteins in trans to build a virus particle containing complementary RNA for a foreign DNA. For example, plasmids with the coding sequences for the gag, pol, and env genes plus nonviral upstream promoter/enhancer sequences (P) can be transfected into suitable mammalian cells, where they are stably maintained and where they produce viral structural and enzymatic proteins. When a retroviral vector is introduced into the packaging cell, vector construct RNA can be packaged, resulting in the production of virus particles containing a vector construct RNA genome. This virus can be harvested and then used to infect target cells to introduce foreign gene X in the vector construct into the cells, and to make gene X product. Because these target cells do not express viral proteins, the vector will not be propagated further. (The viral genes in the packaging cells are not carried along with the vector because they lack the cis-acting sequences necessary for propagation.)
Transduction using nonretroviral vectors
Various types of DNA virus have also been used to transduce mammalian cells, nota- bly adenoviruses that normally cause benign infections of the upper respiratory tract in humans. The linear, double-stranded DNA genome remains nonintegrated within the cell nucleus, and unlike retroviruses they are able to infect both dividing cells and
nondividing cells. So, in addition to transducing cultured mammalian cells, they can be used to ferry transgenes into nondividing cells in vivo.
For use as vectors, adenoviruses have two major advantages over retroviruses. First, the large genome size means that adenoviral vectors can accept quite large insert DNAs—
“gutless” adenoviral vectors (which lack any of the adenoviral genes) can accommodate transgenes up to 35 kb in length. (A packaging cell line is required to provide viral proteins in trans, just as in the case of retrovirus vectors.) Second, adenoviruses can be produced in very high titers (much higher than retroviruses) and so they offer high levels of trans- gene expression. Until recently, adenovirus vectors were widely used in gene therapy, but safety concerns have led to the use of alternative vectors, as described in Chapter 22.
Host range and tropism
Viruses gain access to cells after a protein on the outer virus surface recognizes and binds to a specific receptor on the host-cell surface. Different viruses bind different cell surface receptors, and even different strains of the same virus sometimes use different receptors. The virus (and virus vectors) may be limited to infecting cells from one species (the receptor is not so well conserved between species) or may be able to infect cells from a range of species (the receptor may have been very highly conserved during evolution).
In the case of Moloney murine leukemia retroviruses, for example, the surface envelope glycoprotein encoded by the env gene is the protein responsible for receptor binding, but according to the strain of virus, the virus particles may infect murine cells only (the env gene is said to be ecotropic) or a range of mammalian species, including humans.
The tropism extends to cell type. For example, HIV is tropic for certain immune sys- tem cells that bear the CD4 receptor, notably helper T cells, macrophages, and dendritic cells. Certain strains of the same virus may preferentially infect cells of different types, as in the case of strains of adeno-associated virus.
8.2 PRINCIPLES OF TRANSGENE EXPRESSION IN MAMMALIAN CELLS
Transgenes introduced into mammalian cells often have internal sequences that are designed to be expressed. A minimum requirement is to provide some strong upstream pro- moter to drive expression to make an RNA product. If the transgene RNA is intended to be an mRNA, a downstream poly(A) addition signal will have been stitched into the transgene.
If a transgene is intended to be incorporated into the germ line and subsequently be present in the cells of a transgenic animal, suitably tissue-specific or stage-specific promoter regulatory sequences may be stitched into the transgene so that its expression may be designed to occur in particular tissues or at particular developmental stages appropriate for its intended function.
The transgene may be designed to exist in an extrachromosomal location in the cell, or it may be intended to be incorporated into a chromosomal DNA molecule. Integrated transgenes have the advantage that they will be replicated and transmitted to descen- dants of the original cell, but the chromosomal DNA environment may strongly influ- ence the extent to which the transgene is expressed (as described below).
Inducible promoters allow control over transgene expression
Maximum control over transgene expression is provided by inducible promoters that can be switched on and off according to need, usually by controlling the supply of a par- ticular chemical ligand. Typically, the transcription factors that regulate such promoters are structurally modified by this ligand. Naturally inducible promoters have been used, but more robust inducible promoter systems have been developed either by using non- mammalian, often bacterial, components, or by engineering mammalian proteins so that they are incapable of responding to endogenous inducers. Two widely used systems for regulating inducible transgene expression are described below.
Tetracycline-regulated expression
Here, expression is regulated at the transcriptional level. Two transgenes are involved. One has an E. coli tetR gene to constitutively express the Tet repressor protein. The other has the sequence of interest (usually a coding DNA) and an upstream promoter with the tetO operator in the intervening space. By specifically binding to the tetO sequence, the Tet repressor protein blocks expression of the downstream coding sequence. Gene expression can be restored, however, by providing doxycycline, a tetracycline analog that binds to the Tet repressor causing it to change its conformation and be removed from the tetO sequence (Figure 8.10A). Because expression is induced at the transcriptional level rather than at the protein level, a significant delay may occur before expression is induced in response to providing or removing the induction signal.
transgene A tetO
TetR
P
P
P
P tetO transgene A tetR
P P ER TG
A. B.
transgene A transgene OFF
transgene ON tetO
TetR
transgene A tetO
doxycycline tamoxifen
TetR
TetR
ER TG ER TG
ER TG active
inactive Hsp90
Figure 8.10 Inducible expression of transgenes. (A) Tetracycline-inducible expression. A tetO operator inserted just upstream of transgene A is a recognition sequence that can be specifically recognized and bound by the E. coli Tet repressor protein (TetR). When constitutively expressed from a separately introduced tetR transgene, the Tet repressor protein binds to the tetO operator, and so prevents the expression of transgene A. However, if doxycycline (an analog of tetracycline) is subsequently provided, it binds to the Tet repressor protein, causing it to change its conformation so that it is no longer capable of binding the tetO operator.
As a result, expression of the previously silenced transgene A is switched on. Note that toxicity effects reflecting high-level constitutive expression of the repressor limit the use of this system in some cells.
(B) Tamoxifen-inducible expression. Here, coding sequences for the ligand-binding domain of a mutant mouse estrogen receptor (ER) are fused to a transgene of interest (TG) in an expression vector. The expressed ER–TG fusion protein is bound by the Hsp90 inhibitory protein complex, preventing it from performing its function.
The mutant ER ligand-binding domain is not recognized by estrogen, but will bind to tamoxifen, an estrogen analog. Following tamoxifen binding, the fusion protein is released from the Hsp90 complex, and the protein of interest is activated, even though it is part of a fusion protein. P, promoter.
Tamoxifen-regulated expression
Where rapid induction and rapid decay of gene expression are essential, an induc- ible system that works at the protein level is needed. The estrogen receptor is normally bound by the Hsp90 protein inhibitory complex, keeping it inactive within the cyto- plasm until its ligand, estrogen, enters the cell. Estrogen binds to the C-terminal ligand- binding domain of the estrogen receptor, causing a change of conformation. As a result, the Hsp90 inhibitor is released and the activated estrogen receptor translocates to the nucleus to activate certain target genes (Figures 3.2 and 3.3 give the structure of nuclear hormone receptors and the mechanism of activation by ligands).
Tamoxifen-inducible expression uses a mutant ligand-binding domain of the mouse estrogen receptor, Esr1. The mutant domain does not bind its natural ligand estrogen at physiological concentrations, but will bind the synthetic ligand 4-hydroxytamoxifen.
A cDNA for the mutant Esr1 ligand-binding domain is fused to the coding sequence of a gene of interest. Hsp90 binding prevents expression of the fusion protein until tamoxifen is provided (Figure 8.10B).
Transient and stable expression in mammalian cells
Recombinant DNA clones containing a mammalian cDNA with appropriate expression signals can be expressed in bacterial cells to give high-level expression of mammalian proteins, but the biological properties of the expressed proteins may not be fully repre- sentative of the native molecules. Expressing mammalian proteins in mammalian cells has the obvious advantages that correct protein folding and post-translational modifi- cation are generally not an issue, and it is possible to analyze downstream signals and cellular effects. Once transfected into mammalian cells, a transgene with appropriate expression signals can be expressed for short or long periods, according to the expres- sion systems used, as listed below.
• Transient expression. Here, the transgene remains as an independent, extrachro- mosomal genetic element (an episome) within the transfected cells. In cultured cells, expression of the introduced gene often reaches a maximal level about two or three days after transfection of the expression vector into the mammalian cell line, but that may be sufficient to carry out certain types of gene analysis.
Thereafter, expression can diminish rapidly as a result of cell death or loss of the expression construct. (But in long-lived, nondividing cells, expression of non- integrated transgenes may be maintained for long periods.)
• Stable expression. Here, the aim is to have the transgene integrate into the host cell’s genome. That often requires a virus vector (retrovirus vectors are highly efficient at integrating into chromosomal DNA) but plasmid expression vectors can also randomly integrate at low frequencies, and stable cell lines can be developed with an integrated
gene of interest. The advantage here is that if the transgene has integrated so that the introduced gene is expressed, all descendant cells will contain this gene and expres- sion can be maintained over many cell generations. Neighboring inhibitory regula- tory elements and closed chromatin structure may result in silencing of the transgene (“position effects”). The structure of the transgenic locus may also have an effect when more than one copy of the transgene inserts at the same chromosomal location.
Transient expression: the example of COS cells
Stable expression systems in mammalian