I spent many late nights in the lab with Junwon - I'm so thankful you were there... every night... otherwise no one would be around to criticize the lack of purity of my NMRs. I would like to thank Agnes Tong for organizing things and coordinating events in the department. Significant research efforts in the field of transition metal catalysis have led to the development of powerful methods for C–C and C–heteroatom bond formation.
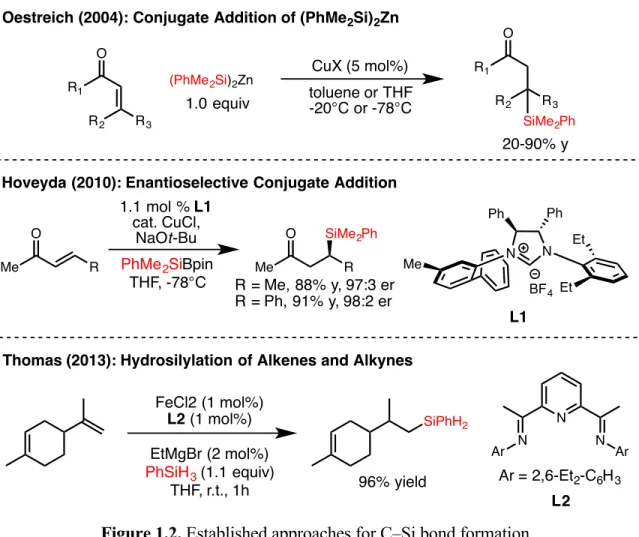
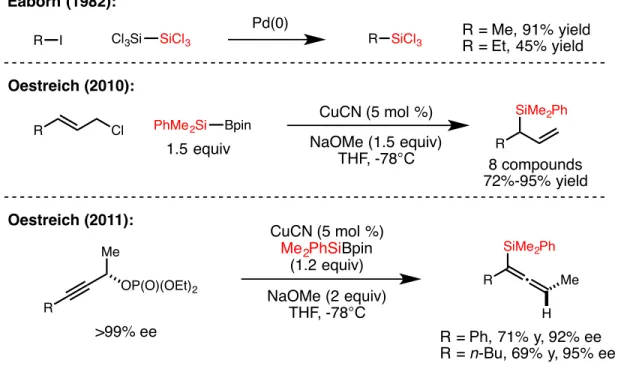
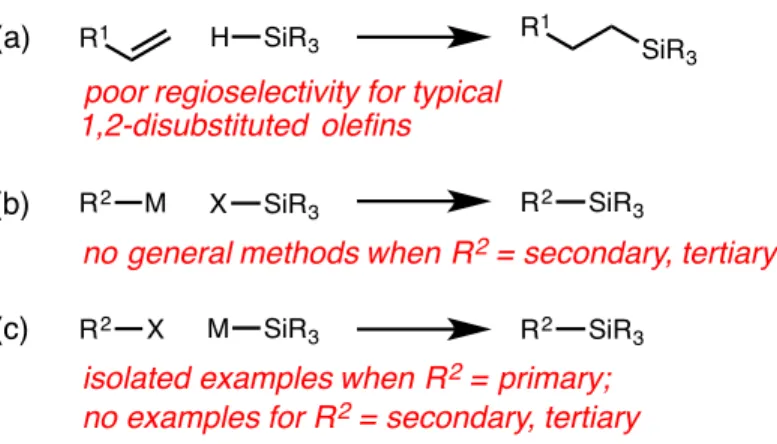
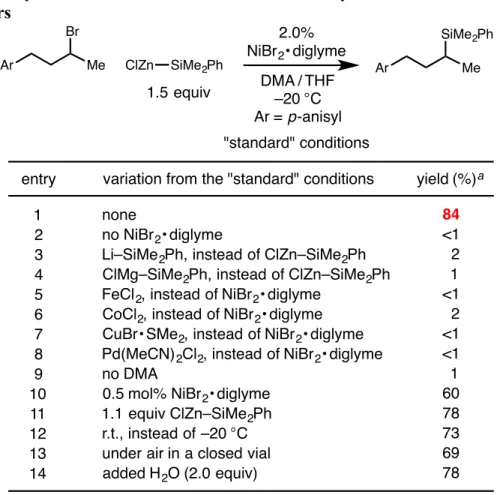
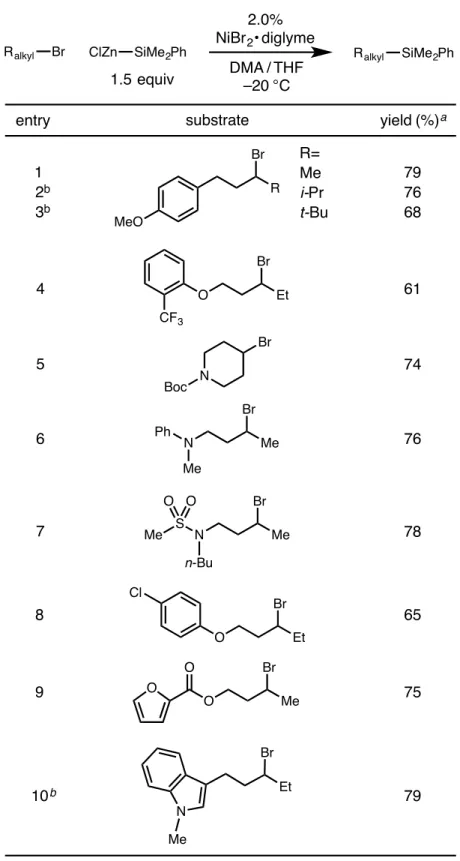
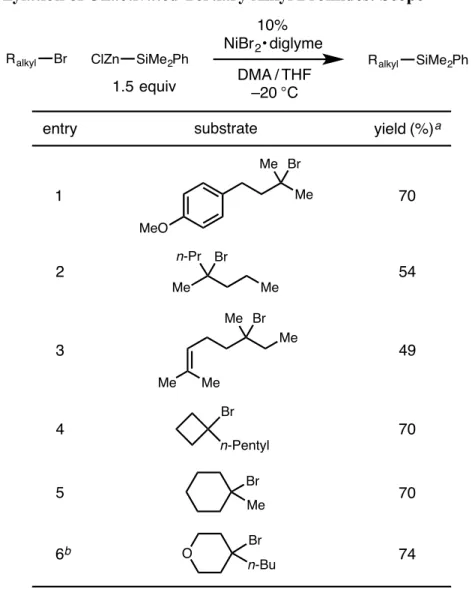
Although this class of reactions was mainly applied to the formation of C–C bonds, the extension of this strategy to the formation of C–B bonds presented by Fu and colleagues inspired the work presented in the second chapter of this thesis , which contains details. the development of a nickel-catalyzed cross-coupling reaction of unactivated alkyl bromides and silylzinc nucleophiles, leading to C-Si bond formation. In the third chapter of this thesis, the nitrite-modified Wacker oxidation of allyl fluorides to selectively produce b-fluorinated. The Tsuji-Wacker oxidation is a widely used reaction in the laboratory environment for the conversion of terminal olefins to methyl ketones.24 However, although oxidation of terminal olefins is typically expected to proceed to form methyl ketones in accordance with Markovnikov's rule, the presence of proximal functional groups can lead to poor regioselectivity of oxidation.
Ketone-selective Wacker oxidation reported by Sigman
More recently, methods have been developed that promote the selective oxidation of terminal olefins bearing substituents with a variety of electronic properties. In 2009, Sigman and colleagues developed the ketone-selective peroxide-mediated oxidation of terminal olefins enabled by a palladium catalyst carrying a bidentate Quinox ligand (Scheme 1.1).25. This system provides high ketone yields for a wide variety of protected allyl alcohols and simple olefins.
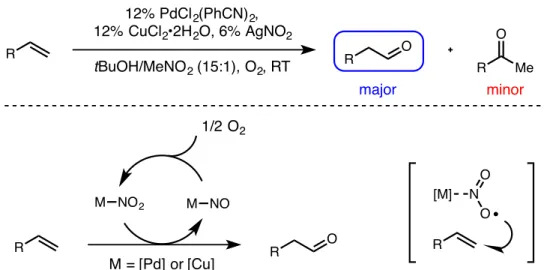
Compared to ketone-selective oxidations, the development of an aldehyde-selective Wacker oxidation has proved more elusive. In recent years, work by the Grubbs26 and Feringa27 groups has demonstrated aldehyde selectivity in the presence of a wide range of functional groups. This work is inspired by preliminary work reported by Feringa in the 1980s, in which a palladium nitrite catalyst offers modest aldehyde selectivity when using tert-butanol as solvent (Scheme 1.2).28 However, this reaction was limited by a low oxidation yield.
Aldehyde-selective Wacker oxidation reported by Feringa
After warming to room temperature, the mixture was filtered under a nitrogen atmosphere by injecting it through a syringe filter directly into a nitrogen-filled 20 mL scintillation vial sealed with a septum cap. After warming to room temperature, the vial was transferred to the glove compartment and the reaction mixture was filtered by injecting it through a syringe filter. The title compound was synthesized according to the general procedure using 5.0 mol% NiBr 2 x diglyme from 1-(3-bromo-4-methylpentyl)-4-methoxybenzene (190 mg, 0.70 mmol).
The title compound was synthesized according to the general procedure using 5.0 mol% NiBr2 × diglyme from 1-(3-bromo-4,4-dimethylpentyl)-4-methoxybenzene (200 mg, 0.70 mmol). The title compound was synthesized according to the general procedure using 2.0 mol% NiBr2 × diglyme from 1-((3-bromopentyl)oxy)-2-(trifluoromethyl)benzene (218 mg, 0.70 mmol). The title compound was synthesized according to the general procedure using 10 mol% NiBr2 × diglyme from 1-(3-bromo-3-methylbutyl)-4-methoxybenzene (180 mg, 0.70 mmol).
The mixture was allowed to warm to r.t., n-tetradecane (26 µL) was added to the vial, and the reaction mixture was analyzed by GC. The mixture was allowed to warm to r.t., n-tetradecane (26 µL) was added to the vial, and the reaction mixture was analyzed by GC.
Strategies toward alkylfluorine compounds
For all reactions in Figure 1: After the work-up procedure, nitrobenzene (0.1 mmol, 10.3 µL) was added as a standard and 1H NMR analysis of the crude product was performed to determine yield and selectivity. The fluoride was prepared according to general procedure A from the corresponding trichloroacetimidate, 5-phenylpent-1-en-3-yl 2,2,2-trichloroacetimidate. The fluoride was prepared by general procedure A from the corresponding trichloroacetimidate, dec-1-en-3-yl 2,2,2-trichloroacetimidate.
The fluoride was prepared according to general procedure A from the corresponding trichloroacetimidate, 1-phenoxybut-3-en-2-yl-2,2,2-trichloroacetimidate. The fluoride was prepared according to general procedure A from the corresponding trichloroacetimidate, 1-(benzyloxy)but-3-en-2-yl-2,2,2-trichloroacetimidate. The fluoride was prepared according to general procedure A from the corresponding trichloroacetimidate, 2-(2,2,2-trichloro-1-iminoethoxy)but-3-en-1-ylbenzoate.
Fluoride was prepared according to General Procedure A from the corresponding trichloroacetimidate, ethyl 6-(2,2,2-trichloro-1-iminoethoxy)oct-7-enoate. Fluorine was prepared according to General Procedure A from the corresponding trichloroacetimidate, 7-chlorohept-1-en-3-yl 2,2,2-trichloroacetimidate. Fluorine was prepared according to General Procedure A from the corresponding trichloroacetimidate, 1-(1,3-dioxoisoindolin-2-yl)but-3-en-2-yl 2,2,2-trichloroacetimidate.
Fluoride was prepared according to General Procedure B from the corresponding alcohol, dec-1-en-4-ol. Fluoride was prepared according to General Procedure B from the corresponding alcohol, dec-1-en-5-ol. Fluorine was prepared according to General Procedure A from the corresponding trichloroacetimidate, 2,2,2-tetradec-1-en-3-yl.
This mixture was injected using an oxygen-filled balloon for ∼60 seconds, and the balloon was left attached to the vial for the remainder of the reaction. 1 H NMR analysis of an aliquot of the crude reaction mixture (without any rotary evaporation step) showed 42:1 aldehyde selectivity and 81% yield. Nitrobenzene (0.1 mmol, 10.3 µL) was added as a standard and 1 H NMR analysis of the crude product was performed to determine yield and selectivity.
Proposed Dissociative Mechanism for Second-Generation Ruthenium Olefin Metathesis Catalysts
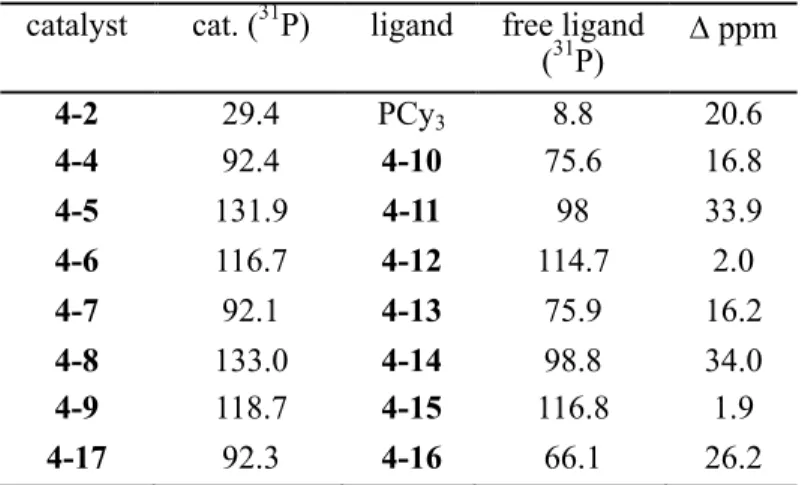
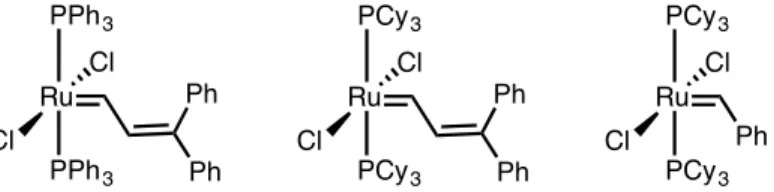
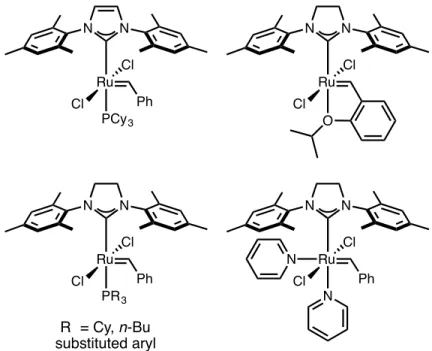
Mechanistic studies of olefin metathesis, promoted by second-generation ruthenium catalysts, have suggested that these reactions proceed by a dissociative pathway, in which phosphine dissociates to form a 14-electron intermediate in an initial step prior to coupling of the olefin (Scheme 4.1). , the activity of these catalysts is affected by the rate of phosphine dissociation (k1; initiation rate) and the relative rate of phosphine recombination (k-1). After formation of the 14-electron intermediate, the probability of phosphine recombination versus productive olefin coupling (k-1/k2) can be determined experimentally; higher selectivity for olefin over phosphine coupling, rather than higher initiation kinetics, has been shown to be the underlying reason for the increased activity of 4-2 compared to 4-1,18. While phosphines containing halogen arenes have been investigated, the incorporation of P-X bonds, where X is an electron-withdrawing heteroatom, has been much less investigated in this context.
Such ligands have been widely used in organometallic chemistry and encompass a wide range of accessible s-donating and p-accepting properties.20 NMR studies of aminophosphines, with structure P(NR)3, have shown reduced s-basicity of these ligands in compared to triphenylphosphine.21 Due to the electronic properties and ease of preparation of aminophosphines, these ligands are particularly suitable to systematically investigate the integration of P-X bonds to increase catalyst activity. The kinetics and computational studies described herein demonstrate the importance of several key factors in promoting phosphine dissociation, facilitating the design of novel ligands for efficient ruthenium olefin metathesis catalysts. In this study, nitrogen-containing heterocycles were systematically introduced instead of the cyclohexyl groups in complex 2 to investigate the effect of P-N bonds on catalyst activity.
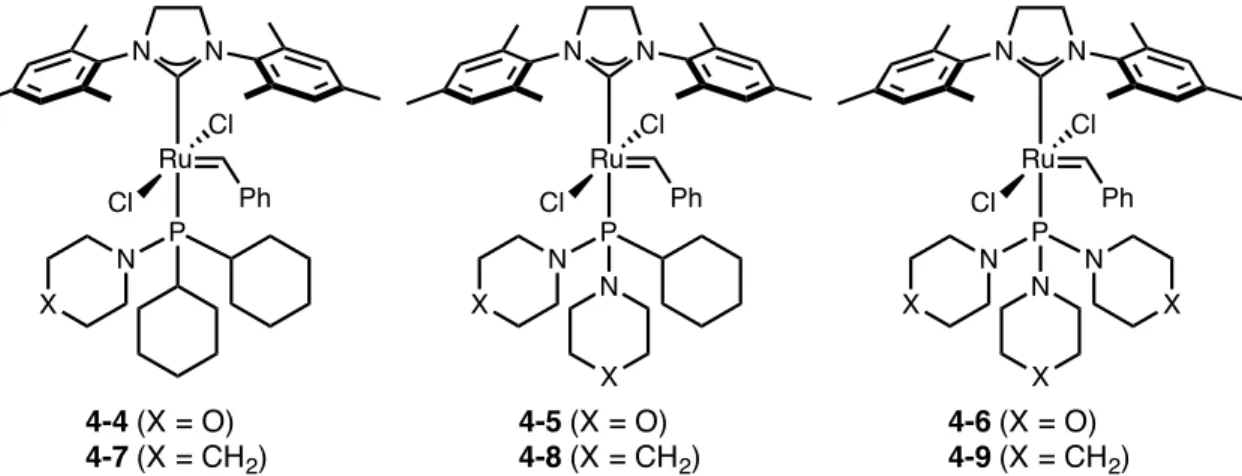
NMR spectroscopic and X-ray crystallographic crystallographic data were obtained to gather structural information, and these data were analyzed in the context of kinetic studies. Moreover, the use of phosphines containing incompatible substituents allows a more complete understanding of the ligand structure, providing additional information about the effects of sterics and ligand conformation on phosphine dissolution. Two new series of second-generation ruthenium olefin metathesis catalysts containing aminophosphine ligands in place of the tricyclohexylphosphine present in catalyst 4-2 were synthesized.
Treatment of the corresponding chlorocyclohexylphosphine or trichlorophosphine starting materials with excess morpholine or piperidine produced the corresponding aminophosphines 4-10 to 4-15 (Scheme 4.2A). After the successful synthesis of the desired ligands, the reaction of bis(pyridine) catalyst 3 with an excess of aminophosphine in THF (Scheme 4.2B), modified from the previously reported procedure, achieved complexation to form catalysts 4-4 to 4-9. 16. Second-generation ruthenium olefin metathesis catalysts with aromatic phosphine ligands are known to initiate faster than their alkylphosphine counterparts, and the effects of replacing the PCy3 ligand with PPh3 in 4-2 and related catalysts have been well studied for ring-closing metathesis ( RCM )22 and ROMP19a reactions.
So after the successful synthesis of catalysts 4-4 through 4-9, we became interested in potentially faster initiating species derived from aromatic amines.
Synthetic Route to Prepare Complexes 4-4 Through 4-9
All catalysts within the piperidine series (4-7 to 4-9) have similar Ru-C1 bond lengths compared to that of the parent catalyst 4-2 (Table 4.2). Under the same conditions, an experiment to determine the initiation rate of catalyst 4-17 resulted in full consumption of the benzylidene faster than the time scale to obtain a precise rate measurement. The rates of phosphine dissociation are reported as the slopes of the lines fitted to pseudo-first-order kinetics; units are (s-1).
In addition, DFT calculations suggest that ligand conformational changes in the transition state of the phosphine dissociation reaction coordinate are responsible for accelerated catalyst initiation rates. The structures were solved by direct methods, successfully locating most of the non-hydrogen atoms. Addition of pentane resulted in the formation of a pink precipitate of the desired complex, 4-4, which was isolated by filtration through celite and dried in vacuo (129 mg, 73%).
Dark brown crystals were obtained by slow Et2O vapor diffusion in a THF solution of the title complex. Addition of pentane resulted in the formation of a pink precipitate of the desired complex, 4-5, which was isolated by filtration through celite and dried under vacuum (79 mg, 67%). Addition of pentane resulted in the formation of a pink precipitate of the desired complex, 4-6, which was isolated by filtration through celite and dried under vacuum (157 mg, 77%).
Addition of pentane led to the formation of a pink precipitate of the desired complex, 4-7, which was isolated by filtration through celite and dried in vacuo (93 mg, 80%). Dark brown crystals were obtained by slow diffusion of Et2O vapor into a benzene solution of the title complex. Addition of pentane led to the formation of a pink precipitate of the desired complex, 4-8, which was isolated by filtration through celite and dried in vacuo (82 mg, 54%).
Addition of pentane resulted in the formation of a pink precipitate of the desired complex, 4-9, which was isolated by filtration through celite and dried under vacuum (92 mg, 78%). Dark brown crystals were obtained by slow diffusion of pentane vapor into a THF solution of the title complex. Addition of pentane resulted in the formation of a pink precipitate of the desired complex, 4-17, which was isolated by filtration through celite and dried under vacuum (110 mg, 96%).