Introduction
Cross-Couplings with Radical Intermediates
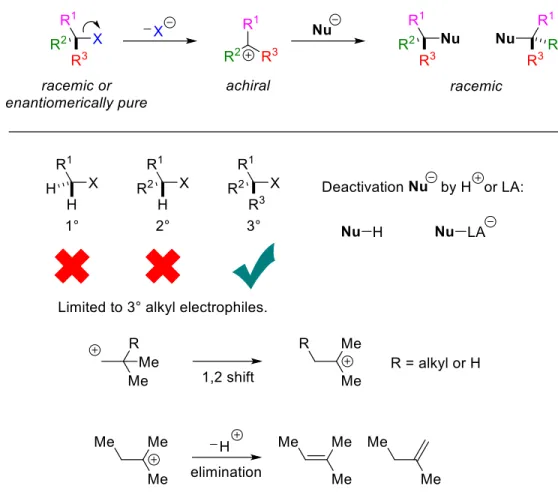
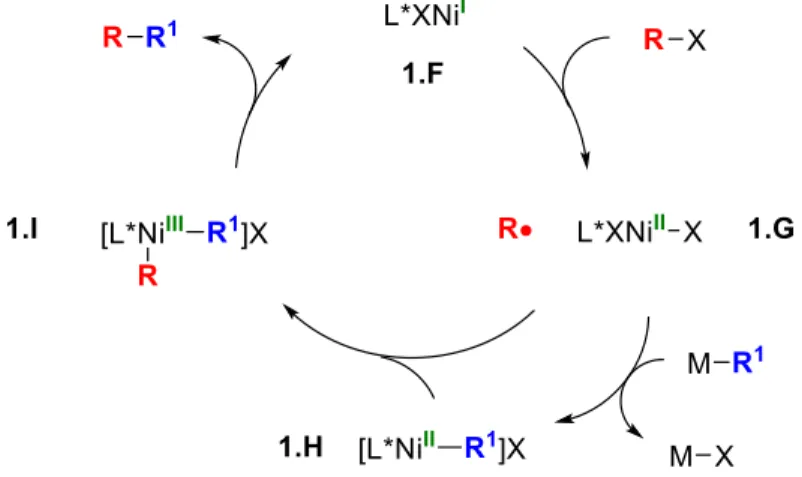
General overview of a transition metal-catalyzed cross-coupling reaction of an aryl electrophile with a generic nucleophile via two-electron oxidative addition. Fortunately, there are alternatives to the two-electron oxidative addition pathway that allow for the net oxidative addition of alkyl electrophiles.7 Transition metals can undergo a two-step oxidative addition by first generating an alkyl radical (via abstraction of halide atoms, single electron transfer, or a related process). ) followed by recombination of the radical and .
Photoinduced Copper Catalysis
The Fu and Peters groups have shown that the copper complex 1.3 used to catalyze the alkylation of carbamates under visible light irradiation (eq 1.3) can undergo photoexcitation and its excited state quenched by an alkyl bromide—probably by a single electron transfer event. 13 l. Based on the previous studies of the Fu and Peters groups, a general mechanism (shown in Figure 1.6) can be considered in which the copper(I)-nucleophile complex (1.A) can undergo photoexcitation to 1.B.

Nickel Catalysis
Fu's group has been very interested in enantioconvergent nickel-catalyzed alkyl-alkyl cross-couplings for many years. Chapter 5 of this dissertation discusses the development of a nickel-catalyzed reaction using a racemic electrophile and an achiral nucleophile (Figure 1.8, top).
Notes and References
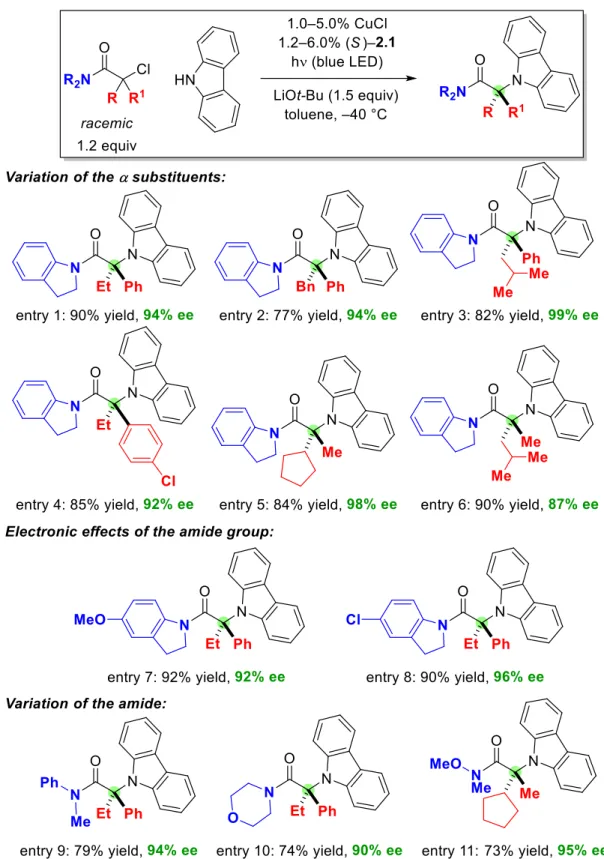
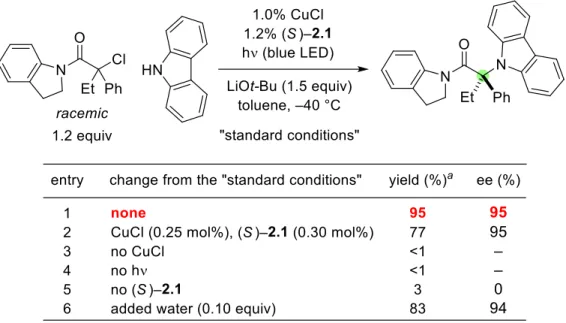
With respect to the nucleophilic coupling partner, substituted carbazoles are also suitable substrates (Figure 2.3, entries 1–5); The enantioconvergent C–N cross-coupling could be performed on a gram scale with a similar result (entry 2: 1.29 g of product, 94% yield, 94% ee). The mixture was then allowed to cool to r.t., the precipitate was removed by filtration and the solvent was removed by evaporation.
Asymmetric Copper-Catalyzed C−N Cross-Couplings Induced
Introduction
We have been interested in photocatalytic approaches to C–N10 bond construction given the high value of amines in fields ranging from biology to chemistry and materials science.11 While initial efforts to develop transition metal-catalyzed C–N cross-coupling reactions, focused on the use of aryl and alkenyl halides as electrophilic coupling partners,12,13 in the last few years, alkyl halides, which are not suitable substrates for classical SN2 reactions, have emerged as useful coupling partners under the combined action of light and copper catalysis.14,15 Until now no development progress has yet been reported. Although significant progress has recently been reported in the development of enantioconvergent cross-couplings of racemic secondary alkyl electrophiles with carbon.

Results and Discussion
- Optimization
- Scope of Reactivity
- Mechanistic Insights
To gain insight into the compatibility of different functional groups with these conditions for enantioconvergent C-N cross-couplings of tertiary alkyl halides, we investigated the impact of additives (1.0 equivalent) on the course of the coupling process, shown in Figure 2.2. 1. To understand whether kinetic resolution occurred, we measured the ee of the unreacted tertiary alkyl halide at the end of the cross-coupling, shown in Figure 2.2, item 1.
Conclusions
This work stands at a previously unexplored intersection of asymmetric synthesis, catalysis with rich-earth metals, photoinduced processes, and cross-coupling reactions of alkyl electrophiles, each of which represents an important current theme in chemical synthesis. We expect our observations to comprise the initial advances in a fertile area of asymmetric catalysis: the enantioconvergent synthesis of secondary and tertiary C heteroatom bonds through photoinduced transition metal-catalyzed couplings of alkyl halides.
Experimental Section
- General Information
- Preparation of Electrophiles
- Asymmetric Photoinduced, Copper-Catalyzed C−N Cross-
- Additive Effects
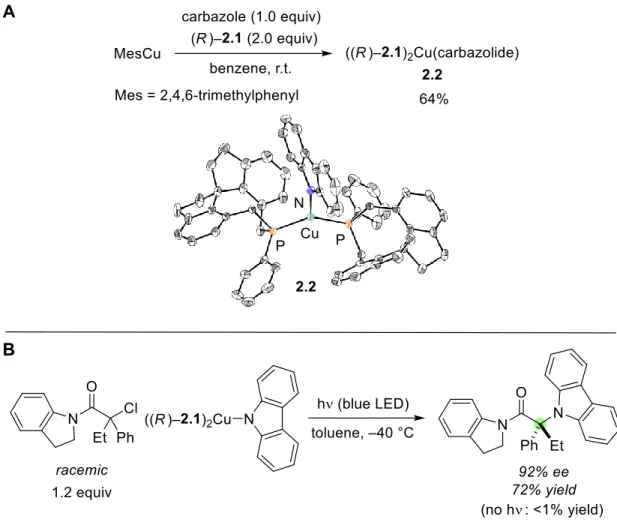
- Synthesis and Reactivity of ((R)−2.1) 2 Cu(carbazolide)
- Determination of Absolute Stereochemistry
Then the catalyst (1.0 mL of the stock solution) was added, and the reaction mixture was stirred at r.t. The resulting solution was concentrated under vacuum, and the residue was purified by flash chromatography Et2O/hexanes), affording 1.29 g (94% yield, 94% ee) of a colorless powder.

Notes and References
Photograph of the reaction setup for visible light-induced copper-catalyzed alkylation of aliphatic amines. For each additive, four reactions were performed: two for GC analysis (to determine the amount of residual additive) and two for 1H NMR analysis (to determine product yield). In particular, the stereocontrolled synthesis of α-chiral amines is of high value, due to their ubiquitous presence in biological systems and among medically important compounds.1 Most methods to access α-chiral amines rely on the application of unsaturated prochiral precursors.2 The most prominent approaches belonging to this group are additions or reductions of imines and enamides3 and hydroaminations4 of olefins.
Due to the heterogeneity of the reaction mixture, we have not been able to perform meaningful kinetic studies.
Copper-Catalyzed Alkylation of Aliphatic Amines Induced by
Introduction
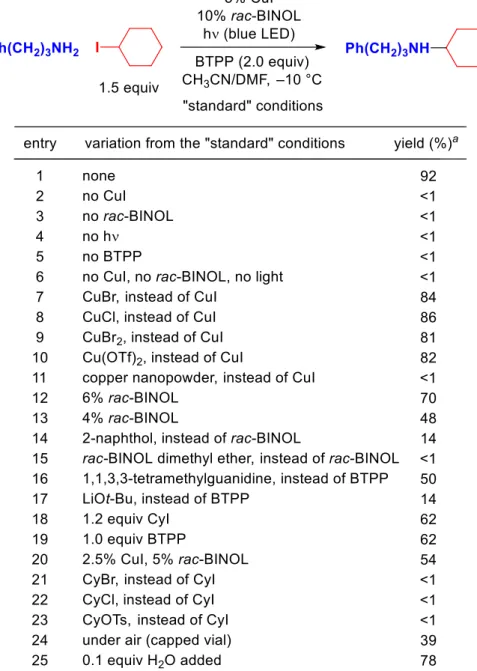
While investigating the functional group compatibility of a method we had developed for photoinduced, copper-catalyzed arylations of phenols,9d we found that the presence of 1.0 equiv of an aliphatic amine additive unexpectedly leads to predominant N-arylation of the aliphatic amine, instead of O-arylation of the phenol (equation 3.4; in the absence of n-BuNH2: 80% yield of PhO–Ar). One of the possible pathways by which phenol could enable the photoinduced copper-catalyzed cross-coupling of an aliphatic amine is shown in Figure 3.2. Ligand exchange of the copper(II) phenoxide with an amine (NH2R) leads to a copper(II)-.
Simplified outline of one of the possible pathways for the photoinduced, copper-catalyzed coupling of an aliphatic amine in the presence of a phenol.

Results and Discussion
- Optimization
- Scope of Reactivity
- Mechanistic Insights
Thus, the efficiency of C–N bond formation does not appear to be very sensitive to the steric demand of the aliphatic amine (entries 1 and 2). If desired, N-protection of the secondary amine can be performed in situ in good yield. For example, after completion of the alkylation shown in entry 1 of Table 3.2, direct trifluoroacetylation followed by purification provides the TFA-protected amine in 86% yield.
Similarly, a 73% yield of the purified carbamate can be obtained after in situ protection with Boc2O.
Conclusions
Development of an Asymmetric Variant
Minor modifications of the conditions shown in Table 3.1 enabled an acyclic secondary alkyl iodide that was previously applicable in the racemic variant (Table 3.2, entry 3) to undergo cross-coupling with 3-phenylpropylamine with modest ee (Table 3.4 , entry 1; 50% yield, 28% ee). Selected examples of ligands screened for enantioinduction in copper-catalyzed asymmetric alkylation of aliphatic amines. Ligand screened for enantioinduction in copper-catalyzed asymmetric alkylation of aliphatic amines using a copper/phosphine photocatalyst.
To this end, a number of ligands were screened in the presence of a catalytic amount of (R)-3.6 (Figure 3.5).
Experimental Section
- General Information
- Preparation of Electrophiles
- Reactions with Neopentyl Iodide
- TEMPO Trapping Experiments
- Additive Effects
- N-Protection of the Amine
- Data on S N 2 Reactions
- Asymmetric Reaction Procedure and Analysis
The reaction mixture is then poured into hexanes (~3 times the volume of the reaction mixture) and filtered. The reaction mixture was then allowed to cool to room temperature and poured into a mixture of CH2Cl2 (25 mL) and a saturated solution of NaHCO3 (25 mL). The reaction mixture was then transferred to a 250 mL round bottom flask with the aid of CH2Cl2, silica gel was added to the flask and.
After the N-alkylation was complete, the reaction mixture was allowed to warm to room temperature.

Notes and References
This transformation is particularly sensitive to the solubility and strength of the base additive (entry 15−18). These results show that the stereoselectivity of the reaction is predominantly controlled by the chiral catalyst. The racemic mixture of the title products was prepared in the same way as the procedure described in the literature20 (route B, scheme 4.1).
HPLC analysis of the product: Diacel CHIRALPAK® IA column; 20% i-PrOH in hexanes, 1.0 ml/min flow rate;.
Asymmetric Copper-Catalyzed Alkylation of N-Heterocycles
Introduction
Amines can be found in a multitude of natural products, pharmaceuticals, agrochemicals and polymers.1 Their prevalence and importance in the chemical world is underlined by a wide range of methods to access these compounds, in particular, through the formation of carbon nitrogen. the bonds. Transition metal catalysis has been a powerful tool for building carbon-nitrogen bonds. However, in contrast to well-established approaches to construct Csp2-N bonds between aryl and alkenyl halogens with amines,6 analogous Csp3-N bond formation . This is in contrast to our previous copper-catalyzed stereoconvergent amine alkylation, where visible light irradiation was essential for coupling.
In addition, the reaction shown in Equation 4.4 provides α-aminolactams, which are common motifs in many bioactive molecules.
Results and Discussion
- Optimization
- Scope of Reactivity
- Mechanistic Insights
Next, we turned our attention to exploring the field with respect to the nucleophilic coupling partner (Table 4.3). It was observed that the ee of the unreacted alkyl bromide increases throughout the reaction, indicating that this electrophile undergoes a kinetic resolution (see Figure 4.4 in the Experimental Section). Our observation that the alkyl iodide illustrated in Table 4.2, entry 1 undergoes an initial increase in ee, which then racemizes during the course of the reaction, suggests that a dynamic kinetic resolution process is operative (see Figure 4.6 in the Experimental Section).
The copper(I) amide complex 4.5 (represented as 4.A) could preferentially react with one enantiomer of the electrophile, resulting in kinetic resolution.

Conclusions
Alternatively, an enantioselective coordinated oxidative addition could occur in which the stereochemistry at the α-position is reversed by an O-linked copper(III) enolate.19 For both the direct SN2 and the oxidative addition/reductive elimination pathway, the copper( I)-amide complex carrying a more electron-rich nucleophile is expected to react faster, but in the latter case only if oxidative addition is slower than reductive elimination.
Experimental Section
- General Information
- Preparation of Electrophiles
- Copper-Catalyzed Asymmetric C sp3 −N Couplings
- Mechanistic Studies
- Determination of Absolute Stereochemistry
After purification by flash chromatography (20→80% Et2O in hexanes) and reverse phase chromatography (0→70% MeOH in H2O), the title compound was isolated as a white solid in 83% yield (66 mg) and 90% ee. After purification by flash chromatography (20→60% Et2O in hexanes) and reverse phase chromatography (0→80% MeOH in H2O), the title compound was isolated as a white solid in 75% yield (67 mg) and 90% ee. After purification by flash chromatography (35→60% Et 2 O in hexanes), the title compound was isolated as a white solid in 54% yield (37 mg) and 83% ee.
After purification by flash chromatography (30 -> 65% Et2O in hexanes), the title compound was isolated as a white solid in 52% yield (38 mg) and 83% ee. After purification by flash chromatography (25 -> 85% Et2O in hexanes), the title compound was isolated as a white solid in 86% yield (69 mg) and 91% ee. After purification by flash chromatography (0->60% Et2O in hexanes), the title compound was isolated as a white solid in 59% yield (43 mg) and 91% ee.

Notes and References
Found in the absence of a copper catalyst, the electrophile reacts with (R)-4.1 in m-xylene at room temperature to form a 1:1 mixture of diastereomers of the corresponding phosphonium salt. When the isolated phosphonium salt (mixture of diastereomers) was used in place of the electrophile in the C–N cross-linking (no catalyst), no product formation was observed. a) Phosphine 4.1 is commercially available from Strem Chemicals. To avoid complications with racemization of the starting material, 3-bromo-1-phenylpyrrolidin-2-one was used in these experiments.
To avoid further complications with the kinetic resolution and the racemization of the starting material, an enantioprene matched 3-bromo-1-phenylpyrrolidin-2-one was used in these experiments.
Enantioconvergent Cross-Couplings of Alkyl Electrophiles: the
Introduction
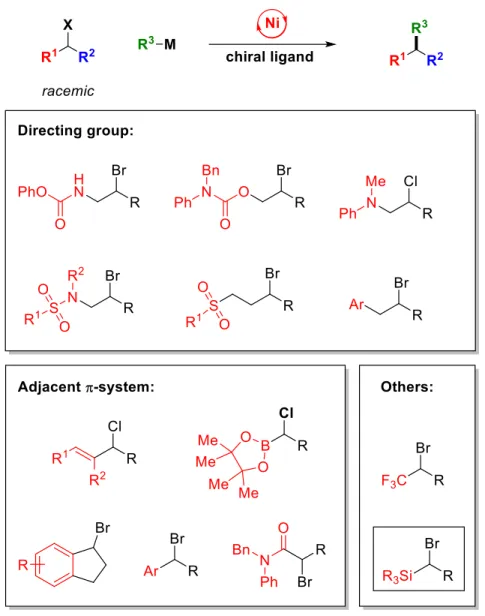
Significant progress has been described in the development of methods for the synthesis of carbon-carbon bonds by enantioconvergent substitution reactions of racemic alkyl electrophiles with carbon nucleophiles.1–3 To date, high enantioselectivity has only been observed in cross-couplings in which the electrophile carries either a directing group (5. I) or a p/π orbital proximal to the leaving group (5.II) (Figure 5.1).4.
Results and Discussion
- Optimization
- Scope of Reactivity
- Mechanistic Insights
Conclusions
Experimental Section
- General Information
- Preparation of (R,R)− or (S,S)−5.3
- Preparation of Nucleophiles
- Preparation of Electrophiles
- Nickel-Catalyzed Alkyl-Alkyl Cross-Couplings
- Enantioenriched Electrophile Experiments
- Determination of Absolute Stereochemistry
Notes and References