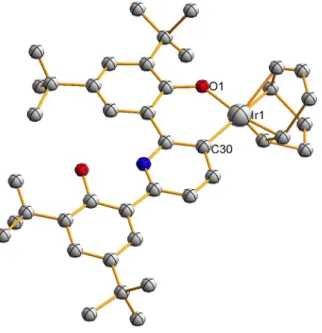
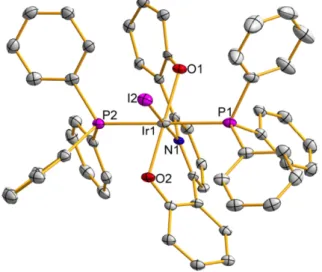
Much of the focus has been on routes involving C−H activation at a transition metal center followed by oxidative cleavage of the resulting C−M bond, offering greater possibilities for good selectivity by avoiding radical-based pathways. The symmetry of the 1H NMR (only two tert-butyl peaks, for example) is consistent with either a five-coordinate structure or a flowing four-coordinate structure, with the phenolates coordinated to Na+ and Ir exchanging rapidly (1 and 1b, respectively, in Scheme 2.1). Second, attempted crystallization of 1 in the presence of 15-crown-5 (attempts to crystallize 1 without 15-crown-5 were unsuccessful) yielded 3 instead, the result of C−H activation at the 3-position of the central pyridine ring (Figure 2.2) .
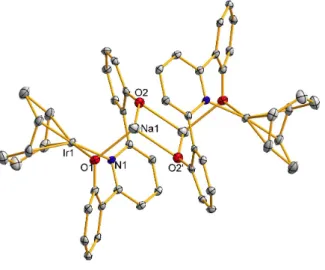
The nearest Na(15-crown-5) is located above the plane of the ligand and has been omitted for clarity. To prevent this unwanted ligand C−H activation, we prepared H2(ONOtBu) with an additional tert-butyl group to inhibit C−H activation of the pyridine linker. The synthesis of the ligand, which was straightforward and analogous to that of H2(ONO), is shown in Scheme 2.2.
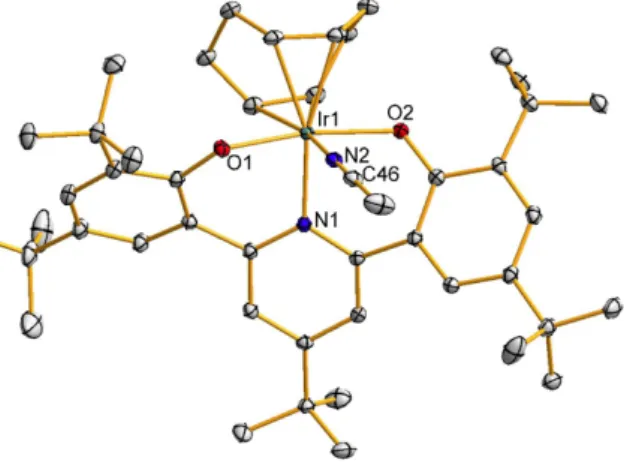
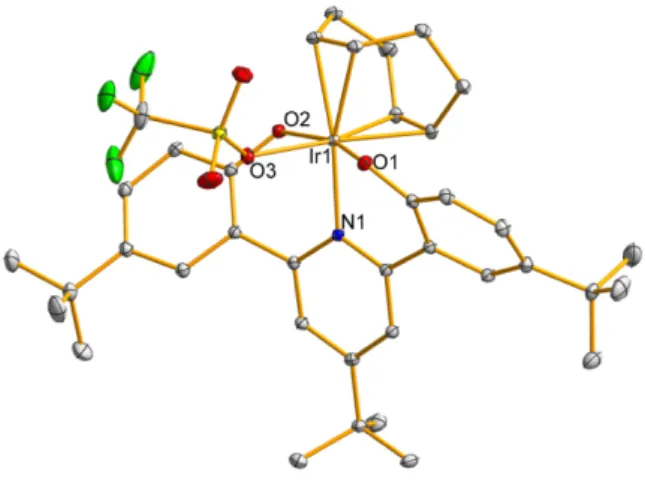
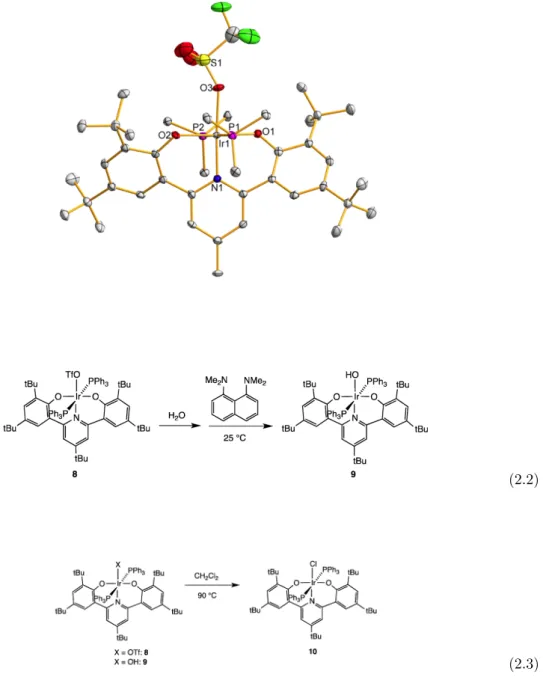
The proposed mechanism for this transformation is one-electron transfer followed by H-atom abstraction (Scheme 2.5); (quantitatively) the shown by-product of the oxidized proton mushroom was observed. In both oxidations of I2, a dark blue color is observed throughout most of the reaction, although all the starting complexes and products are yellow. Reaction of 14 with AgPF6 afforded dark blue [(ONOtBu)Ir(PEt3)2Me]PF6 (16, Scheme 2.7) characterized by a crystal structure and EPR spectrum (Figure 2.15) very similar to those (relatively few) reported organometallic species IrIV [13].
The close similarity of the second potential, in contrast to the first, may indicate that the second wave corresponds to oxidation of the ONOtBu ligand rather than to Ir.).
C−H Activation by Ir III (ONO tBu ) Complexes
All of this suggests that the reaction of 14 (or 12 ) with I2 starts with a single electron transfer to generate an intermediate IrIV -Me cationic species that undergoes SN2 attack by I– on the Ir−C bond. Additionally, the ethyl groups on the phosphine ligands and six benzene molecules per unit cell have been removed for clarity. Thermolysis of 12 in C6D6 yields CH4 as the only detectable isotopologue of methane, and mass spectrometry of the resulting isolated phenyl product 17 shows the incorporation of six D atoms (Eq. 2.5).
A benzene and a pentane solvent molecule, as well as the phosphinephenyl groups, have been removed for clarity. A dodecane solvent molecule and portions of various phenyl and tert-butyl groups have been removed for clarity. These transformations require less than one day for complete conversion (in methylene chloride or p-xylene), comparable to the time required for conversion of 12 to 17 and much faster than the conversion of 17 to 18 in benzene.
The reaction of 12in toluene at 65◦C proceeds in the same way as that in benzene; Although no pure compound could be isolated, the mass spectrum of the mixture is consistent with the formation of an IrIII-tolyl compound, (ONOtBu)Ir(PPh3)2(C6H4CH3). 1H NMR signals would be expected for the (CH2CN) methylene protons, which are diastereotopic in the solid-state structure; only one is observed; this could be due to accidental degeneration or dissociation of the dimeric structure in solution. A benzene solvent molecule, phosphine phenyl and (ONO) tert-butyl groups have been removed for clarity.
The order of product formation (17 and 18 are kinetically and thermodynamically preferred, respectively [15]) seems to suggest that the first C−H activation is the intermolecular reaction with benzene. The H atom leaving with methane clearly comes from a PPh3 ligand and not from solvent benzene, so PPh3 must be the site of the first C−H activation, even though the corresponding product does not appear until much later. Proposed mechanism for the transformation of 12in benzene to the kinetic product17 and the thermodynamic product18.
C-H activation is shown in Scheme 2.8 to proceed via oxidative addition to give an IrV intermediate, a mechanism that has been supported by both experiment [16] and calculations [17]. As a consequence, the solvent C (or aπ-arene adduct analogous to D) has no alternative but to bind PPh3 and 18 forms directly, as it does in solvents (dichloromethane, ether, THF) that are not susceptible to C−H activation at all. The reaction of 14 is much slower, presumably because PEt3 dissociates much less readily and leads only to a phenyl product, indicating that cyclometalation of PEt3 does not occur.
Kinetics of C−H Activation
It seems reasonable that similar considerations apply here as well; but an alternative pathway, such as σ-bond metathesis, cannot be ruled out. In any case, conversion of Ato C should be irreversible, since no multiple H/D exchange is observed, while conversion of CtoF should be reversible, since 17 eventually reverts to 18. Nevertheless, it does: a good exponential fit is achieved (Figure 2.20, left), with kobs s−1 (equivalent to a half-life of about an hour), so any difference in K values is small enough to be neglected .
As expected, the addition of extra L slows down the reaction significantly; a graph of 1/kobs is linear in [L] (Figure 2.21). The rate law for direct conversion of 12 to 18 in p-xylene should be identical to the above, and the reaction exhibits clean first-order kinetics, with a rate (kobs s−1) about a factor of 2 slower than that for 12 to 5. . First, the values of K1 and k2 need not be identical to those in benzene (although they should not be expected to differ very much).
However, any substantial (i.e., sufficient to explain the entire factor of 2) concentration of a new intermediate should have been detectable by NMR. Finally (and perhaps most likely), agreement within a factor of 2 (especially when switching solvents) may be as good as can be expected given the reproducibility considerations discussed earlier. As noted above, the conversion from 17 to 18 is much slower in benzene than in p-xylene, occurring over weeks or days, respectively (Figure 2.22).
This observation is easily explained by Scheme 2.9: the concentration of C, the precursor of 18, will be inversely proportional to the concentration of benzene. The rate law for that mechanism can be derived using the steady-state approximation for both C and F, both of which must be present at low concentrations and in rapid equilibrium with each other. In a benzene solution the reaction must be first order and independent of [L]; both were observed.
We can test this for self-consistency with the observed rate constants, again assuming that there is little difference in parameters between the benzene and p-xylene reactions. Since no direct formation of 18 from 12 was observed, k5 must be small compared to mek4K3[PhH], so the value of kobs is approximately equal to k−4k5/k4K3[PhH]. For the reaction in p-xylene, onset [17] = maximum [PhH] = 8.8 mM and minimum value of k5/k4K3[PhH] = 160, so the assumption required to calculate first-order kinetics is valid how good
![Figure 2.20. Left: kinetics of conversion of 12 to 17 in C 6 D 6 at 65 ◦ C. Right: kinetics of conversion of 12-d 30 ((ONO tBu )Ir[P(C 6 D 5 ) 3 ] 2 Me) to 17-d 36 ((ONO tBu )Ir[P(C 6 D 5 ) 3 ] 2 C 6 D 5 ) in C 6 D 6 at 65 ◦ C.](https://thumb-ap.123doks.com/thumbv2/123dok/10406105.0/23.892.187.794.333.525/figure-left-kinetics-conversion-right-kinetics-conversion-ono.webp)
Conclusions
Supporting Details
Experimental Section
After refluxing for 4 h, an additional 100 mL of hydrogen peroxide (30% in water) was added and the mixture refluxed overnight. Excess POCl3 was removed by distillation, and the remaining liquid was washed with a saturated aqueous solution of sodium carbonate. After refluxing for 4 h under air, an additional 80 mL of hydrogen peroxide (30% in water) was added and the mixture refluxed overnight.
Both solutions were taken out of the box and the tBuLi solution was added to the solution of O-Methoxymethyl-2-bromo-4,6-di-tert-butylphenoxide while the latter was allowed to thaw. 15-Crown-5 (21 µL, 106 µmol) was mixed in and the solution was allowed to sit undisturbed for several days. After stirring for one hour, the solvent was removed in vacuo, redissolved in hexanes and filtered, and the solvent was removed in vacuo again to give 4·THF as a red solid with a glassy appearance.
The headspace was then filled with 2.9 atm H 2 and the NMR tube was continuously inverted to mix. After two hours, the solution was removed in vacuo, redissolved in pentane and filtered, and the solvent was again removed in vacuo. Then the solvent was removed in vacuo and the black solid was redissolved in benzene, filtered and lyophilized to obtain a yellow powder.
A suspension of [FeCp2][PF6] (75.6 mg, 228 μmol) in acetonitrile was added, the solution was allowed to stir overnight, the solvent was removed in vacuo, and the solid was redissolved in benzene, filtered, and lyophilized to afforded 6. A precipitate was immediately observed and the mixture was stirred for four hours, pumped, redissolved in benzene, filtered, and lyophilized, affording 9 (18.8 mg, 14.7 μmol, quantitative yield) as a yellow powder. 9 is slightly soluble in pentane (about 3 mg/mL) and was crystallized by allowing a pentane solution to evaporate. CoCp2 (1.4 mg, 7.4 µmol) dissolved in C6D6 was then added and the entire mixture transferred to a J-Young NMR tube.
The NMR tube was heated to 90◦C and the reaction was followed by NMR; after 26 hours the reaction was complete. Afterwards it was pumped down and the resulting 14 was redissolved in hexanes, filtered to remove by-products and impurities and pumped down again. The residue was redissolved in pentane and filtered, and the solution was pumped down to afford 17 (20.9 mg, 15.6 µmol, 98% yield).
Then it was pumped off, and the resulting 19 was redissolved in hexanes, filtered to remove the by-products and impurities, and pumped off again. In a typical example, 7.8 mg (6.1µmol)12 was dissolved in 700µL C6D6 inside a nitrogen-filled glovebox, and the solution was transferred to a J-Young NMR tube.
Kinetics Details
The specific run is detailed in the corresponding section in Figure 2.23; b) equivalents of PPh3 added;. For PPh3 inhibition experiments, the Ir complex was mixed with different amounts of a 20 mg/mL stock solution of PPh3 in C6D6, and additional C6D6 was added to bring the volume to 700µL.
Crystallographic Data
Acknowledgments
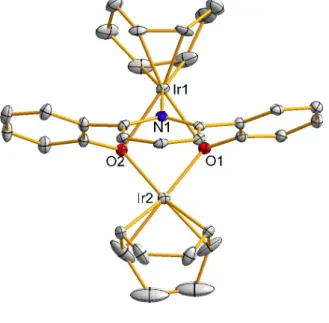
14] The crystal structure included a complex solvent region that could not be satisfactorily modeled and also indicated the presence of Li+ ions, apparently carried over as impurities from an earlier step in the overall synthetic sequence; counteranions required for charge balancing could not be located, probably because they are located within the solvent region.