Uncle Jimmy, thank you for always being there for us and for all the memories as a child. Thanks to Alexia Kim for being a great friend and for all the fun times going to the movies and seeing the cute dogs.
INTRODUCTION
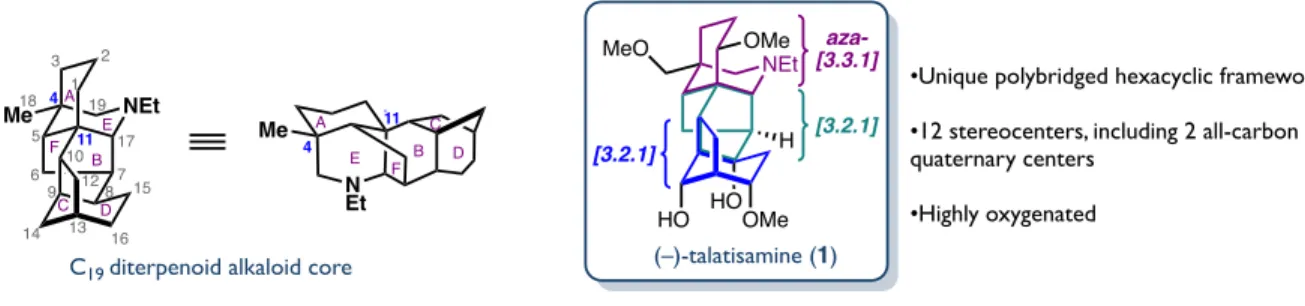
STRUCTURAL ANALYSIS AND BIOSYNTHESIS OF C 19 -DITERPENOID
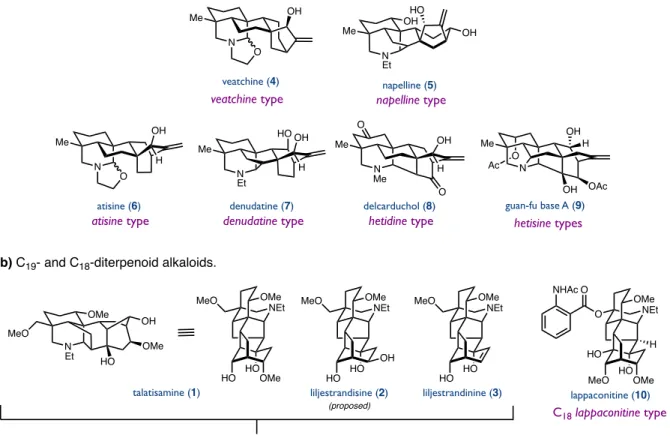
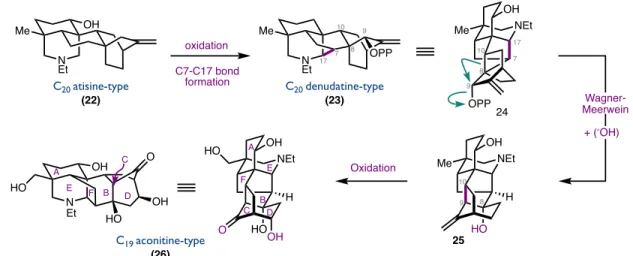
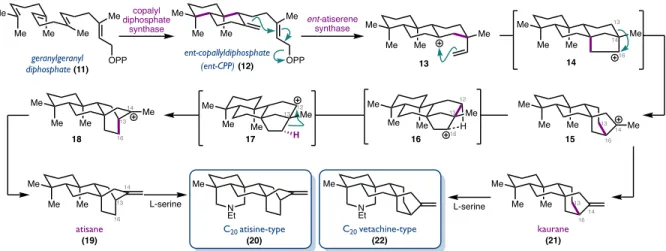
Biosynthetically, diterpenoid alkaloids are derived from the structurally similar families of kaurane and atisane diterpenoids incorporating serine as the nitrogen source.15 These complex skeletons are believed to arise from a series of cationic polyene cyclizations and skeletal rearrangements (Figure 1.316). C19-diterpenoid alkaloids are believed to derive from the Wagner-Meerwein rearrangement of the C20- denudatine nucleus, where [2.2.2]-bicyclooctane is converted to a [3.2.1]-bicyclooctane (Figure 1.4). Cationic rearrangements of carbon skeletons, which are crucial for the biosynthesis of these natural products, have also played an important role in many of the total syntheses of C18- and C19-diterpenoid alkaloids.
TOTAL SYNTHESES OF C 19 -DITERPENOID ALKALOIDS
Biomimetic Wagner-Meerwein Approaches to Aconitine Core
In a subsequent synthesis of the C19-diterpenoid alkaloid 13-deoxydelphonine (39), Wiesner and co-workers attempted to rearrange the denudatin core, where the C7-C17 bond is already in place (Scheme 1.3).19 Starting from the ortho-quinone mono - ketal 34, Diels-Alder cycloaddition with benzyl vinyl ether followed by global reduction with Zn and acetic acid provides the denudatine core in 35. The Sarpong laboratory has used this strategy for the synthesis of a variety of diterpenoid alkaloid carbon skeletons, including aconitine- of the C19-diterpenoid type (Scheme In their elegant 2015 synthesis of lilystradinine (3), they use an intramolecular Diels–Alder addition of an ortho-quinone monoketal with a pendant olefin to construct C/D-bridged [2.2.2]bicycles 42.
Gin’s Synthesis of Neofinaconitine
After elaboration of 48 to diene 49, Diels–Alder fragment coupling with dienophile 50 is performed to form the A-ring of the natural product. The terminal bond of the natural product core is forged by Giese addition of the C7-radical to the D-ring of the enone to give 59.
CONCLUDING REMARKS
An intramolecular Mannich reaction formed the A/E/F-tricycle of the natural product and gave the unexpected product 54 by intramolecular oxy-Michael. This has stimulated many collaborative synthetic efforts, with several groups reporting approaches that convert the carbon framework of one subfamily of natural products to another.
NOTES AND REFERENCES
12
RETROSYNTHETIC ANALYSIS OF C 19 -DITERPENOID ALKALOIDS
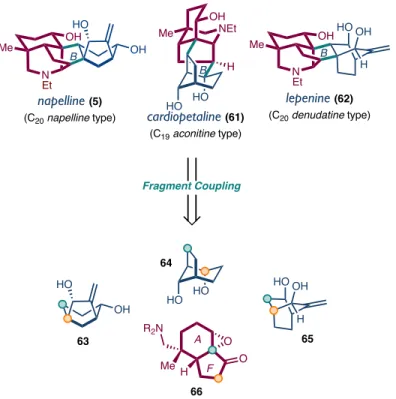
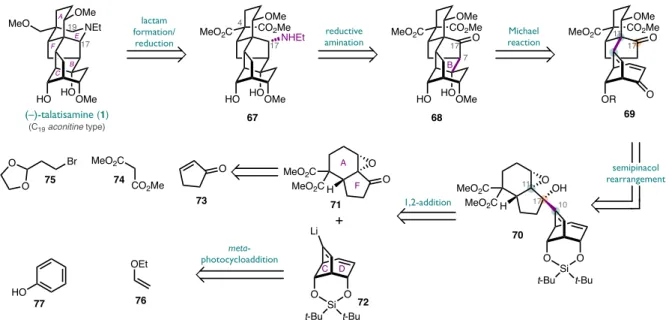
The C19-aconitine, C20-napelline and C20-denudatine type diterpenoid alkaloids share a common A/E/F tricycle linked by two bonds from the central B ring to a C/D cycle characteristic of each subfamily . As such, we envisioned that a retrosynthetic decoupling through the two bonds of the B ring would allow us access to each subfamily through a fragment coupling of the proper C/D cycle with a common A/F cycle .
SYNTHESIS OF FRAGMENT COUPLING PARTNERS
- Synthesis of [3.2.1]-Bicyclooctene C/D-Bicycle
This alkenyl lithium was slowly added via a cannula to a solution of the epoxy ketone 71 at -94 °C. This remarkable two-step sequence brings together all the carbon atoms needed to access the aconitine core and forms the major quaternary C11 center, demonstrating the power of 1,2-addition/semipinacol rearrangement as a fragment-linking tactic.
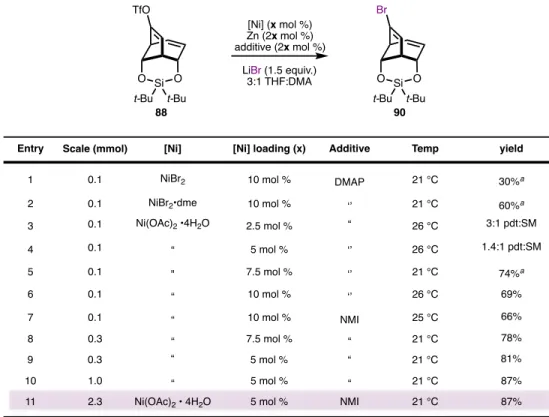
CONCLUDING REMARKS
EXPERIMENTAL SECTION
The mixture was extracted with EtOAc (3 x 1 L), and the combined organic extracts were washed with sat. The mixture was extracted with Et2O (3 x 250 mL), and the combined organic extracts were washed with sat.
NOTES AND REFERENCES
50
SEQUENTIAL REDUCTION OF C18 AND C19 ESTERS
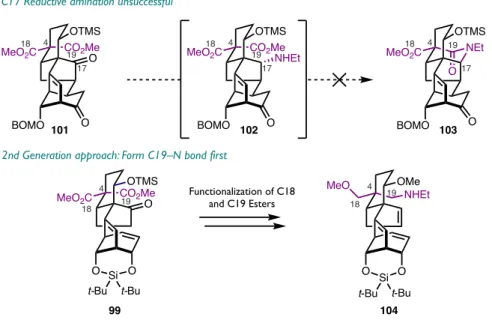
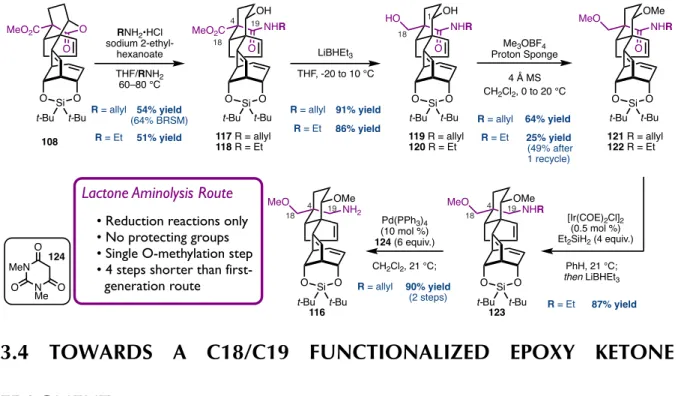
Our first generation approach to form the C19-N bond was to selectively reduce the C18 ester (Scheme 3.1). To differentiate the reactivity of the two esters, the C1 TMS ether of 99 was deprotected with trichloroacetic acid and subsequent treatment with potassium carbonate and methanol in dichloromethane effected lactonization with C19 to yield 106. The C18 ester of 108 can be selectively reduced. to the alcohol using lithium tri-tert-butoxyaluminum hydride, a reducing agent with.
Methylation of the C18 alcohol followed by reduction of the C19 lactone 110 with lithium borohydride gives the C1/C19 diol 111. To introduce the amine at C19, the triethylsilyl ether of 113 is deprotected and the resulting alcohol, 114, is cleanly oxidized to the C19 aldehyde 115 using Stahl's conditions.4 This aldehyde serves as a handle for reductive amination with N-allylamine followed by cleavage of the N-allyl group to give C19 primary amine 116. This route from semipinacol product 99 to primary amine 116 comprises 12 steps, nine of which twelve represents change of functionality at C18 and C19.
SELECTIVE AMINOLYSIS OF C19 LACTONE
TOWARDS A C18/C19 FUNCTIONALIZED EPOXY KETONE FRAGMENT
- Selective Functionalization of a C18/C19 Diol
- Setting C4 Quaternary Center by Michael Addition of a Prochiral
This disparity in the number of steps required to prepare each of the fragments highlights an opportunity to make the synthesis more convergent by preparing a better functionalized epoxy ketone fragment, such as 125 (Figure 3.2). In our workup of the coupling product of fragment 99 with the primary amine 116 , six of the eight required steps were spent modifying C18 and C19. Since these carbons are part of the epoxy ketone fragment 71, if these sites were properly functionalized before coupling the fragment to the synthesis they would be more convergent and therefore more efficient.
We were impressed by this result, as it is a selective protection of the C18 alcohol. We believe that this is due to the instability of the resulting enone 130 in the presence of base. Decreasing the base loading improved the reaction yield with a slight increase in ee.
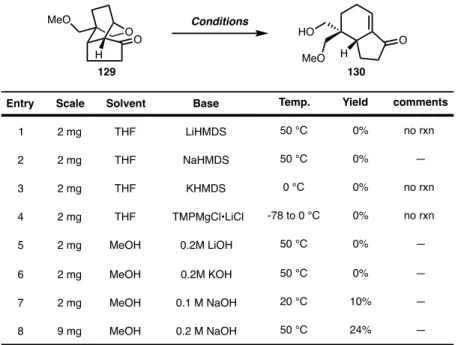
CONCLUDING REMARKS
EXPERIMENTAL SECTION
The resulting crude residue was purified by silica gel chromatography (10% acetone in hexanes to 12.5% acetone in hexanes. The product is highly crystalline, it was loaded into PhMe where some of the product elutes but is collected) to give olefin 108 yield (1.64 g, 3.28 mmol, 93% yield) as a crystalline solid. The reaction was quenched with saturated aqueous 0.1 M NaOH (10 mL), warmed to room temperature and stirred for 20 minutes. The reaction was quenched with saturated aqueous Na 2 SO 3 (1 mL) and the mixture was extracted with EtOAc (4 x 2 mL).
After stirring for 2 h, the reaction was cooled to 0 °C and quenched with saturated aqueous Na 2 S 2 O 3 (100 mL). The reaction was then cooled to room temperature and quenched with saturated aqueous NaHCO 3 (100 mL). After stirring for 30 hours, the reaction mixture was cooled to room temperature before water (80 mL) was added.
After allowing the reaction to cool to room temperature, the mixture was filtered over Celite and the filtrate was concentrated in vacuo. The reaction was quenched with saturated aqueous NH 4 Cl (1 mL) and extracted with EtOAc (5 x 2 mL).
NOTES AND REFERENCES
INTRODUCTION
RADICAL CYCLIZATION CASCADE
- Reaction of Neutral Aminyl Radical
- Reactivity of Aminyl Radical Cations
Starting from N -ethylamide 118 , O -methylation with trimethyloxonium tetrafluoroborate proceeds in moderate yield to afford 175 along with recycled starting material. The siliconide is deprotected with HF•pyridine to provide a diol, which can undergo selective oxidation at the allylic site under Stahl's conditions. The radical then undergoes Giese addition to the D-ring enone at C6 to afford the observed product 179 after quenching of the resulting α-radical.
To quickly evaluate the conditions for radical cyclization to form the E-ring piperidine, we turned to a simplified model system. A small series of single-electron reducing metal reagents were tested on our model substrate 197 to see whether 6-exo-trig cyclization could occur to yield the E-ring piperidine (Table 4.1). Previous synthetic work by Gin and colleagues, as well as work from our laboratory (vide infra) suggests that a C7 radical should readily cyclize with a D-ring enone to close the central B-ring (Figure 4.3).7 The absence of C7– Formation of C8 bonds under copper(I) catalyzed conditions.
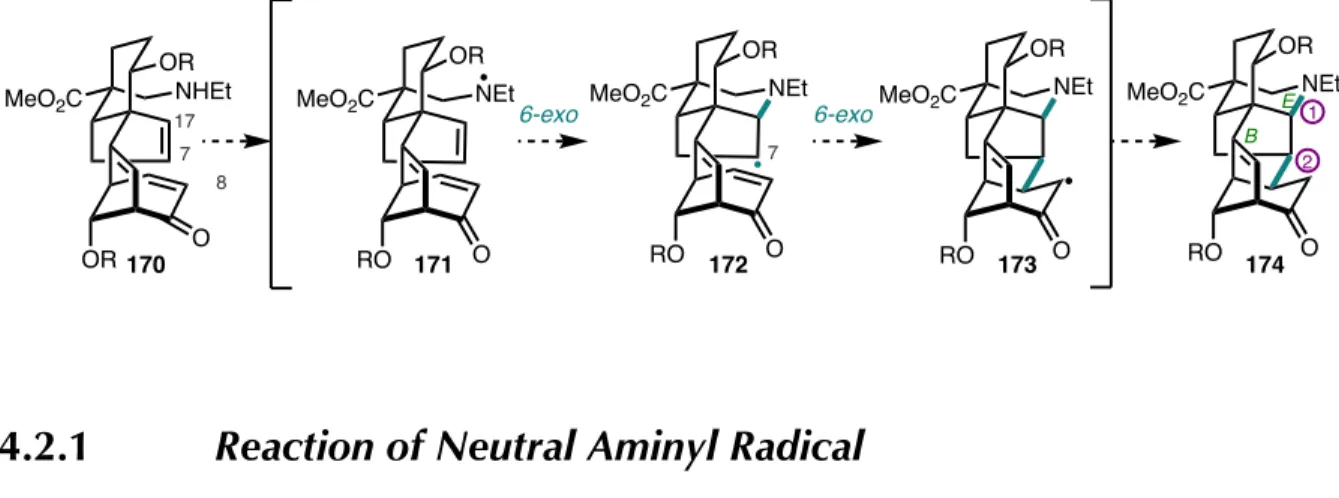
SYNTHESIS OF C 19 -DITERPENOID ALKALOIDS (–)-TALATISAMINE,
- Completion of the Aconitine Core
- Reduction/Oxidation of D-ring for Divergent Syntheses of (–)-
In addition to forming the C17-N bond of the aconitine core, this strategy allows for the functionalization of C7. To form the final C7–C8 bond of the aconitin core, we envisioned a Giese addition of a C7 radical generated from 230 to a D-ring enone. Having demonstrated retrosynthetic uncoupling through the central B ring as a viable strategy to access the aconitine core, reduction/oxidation manipulations of the D ring were the only remaining challenges to access natural products 1 , 238 , and 3 .
Lithium aluminum hydride and sodium borohydride each favored the formation of the axial alcohol 235 , while samarium(II) iodide favored the 2:1 equatorial alcohol 236 in excellent yield. Deprotection of the C14 MOM ether of 237 gave a product with a 1H NMR spectrum that differed significantly from that of the material isolated from nature.10 The reported structure 2 represents the only reported natural product C19-diterpenoid alkaloid with the stereochemistry of the equatorial alcohol at C16 . As such, we hypothesized that the reported structure of lillestrandisin is epimeric at C16 of the actual natural product structure.
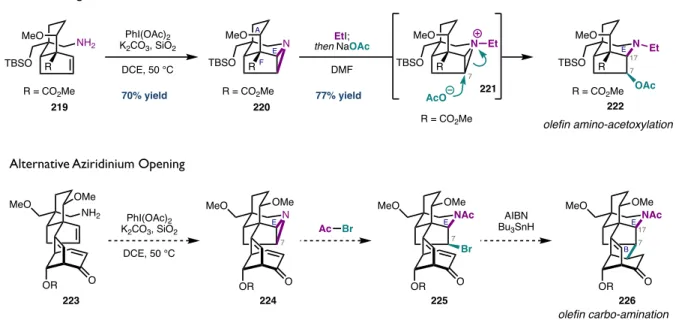
CONCLUDING REMARKS
232 is treated again with Mukayama's reagent, but now quenched with methanol and pyridine to give the C8 methyl ether. The ketone C16 is converted to the corresponding enol triflate, then reduced under palladium-catalyzed conditions to afford 240. Reduction of the amide with lithium aluminum hydride followed by heating to 110 °C in aqueous sulfuric acid results in deprotection of the C14 MOM ether and substitution of SN1 with water at C8 to give (–)-lillestrandinine (3) in 60% yield over 2 steps.
We believe that the success of this new approach shows that there are still opportunities for strategic innovation to access these highly related natural product scaffolds. The synthesis could be more convergent if the epoxide ketone coupling partner bearing these centers had installed the appropriate functionality prior to the coupling event. We will seek to apply our fragment coupling strategy to the synthesis of other subfamilies of diterpenoid alkaloids, C20 denudatin and C20 napeline-type natural products in particular, by varying the bridging bicyclic coupling partner.
EXPERIMENTAL SECTIONS
The reaction was then cooled to room temperature, loaded onto a silica gel plug, which was The reaction was allowed to stir for an additional 10 min at −78 °C, then quenched. The reaction was allowed to stir for an additional 10 minutes at 0 °C, then the reaction was quenched.
The reaction was quenched with saturated aqueous NaHCO 3 (50 mL) and the mixture was allowed to warm to room temperature. The reaction was allowed to stir at 80°C for 2 days and the progress of the reaction was monitored. The reaction mixture was stirred for 90 min before filtering over a short pad of silica gel eluting with 1:1 hexanes:EtOAc.
The reaction mixture was stirred for 30 min before filtering over a short pad of silica gel eluting with EtOAc. After the addition of the AIBN/n-Bu3SnH solution was completed, the reaction mixture was cooled to room temperature and concentrated in vacuo.
NOTES AND REFERENCES