Denise Grünenfelder was one of my closest lab friends here at Caltech. Some of my other close friends in the lab who I will miss greatly: Drs.
INTRODUCTION
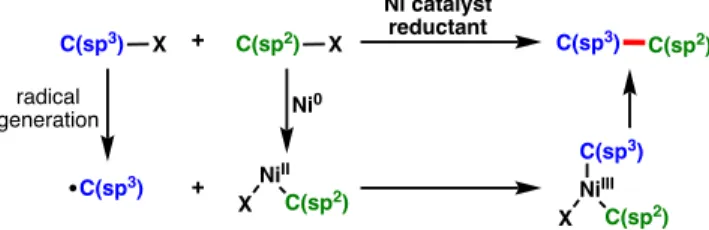
Towards this effort, a number of examples of stereoconvergent and stereospecific Ni-catalyzed cross-coupling reactions using C(sp3)-hybridized electrophiles have been developed (Figure 1.2). Here, we discuss the development of Ni-catalyzed reductive cross-coupling reactions using C(sp3)-hybridized electrophiles.
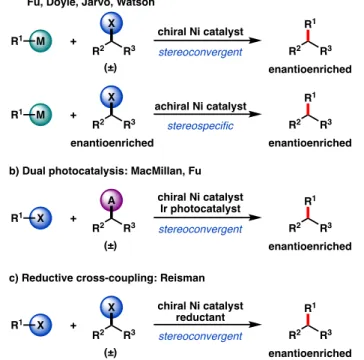
CROSS-COUPLINGS WITH HALIDE ELECTROPHILES
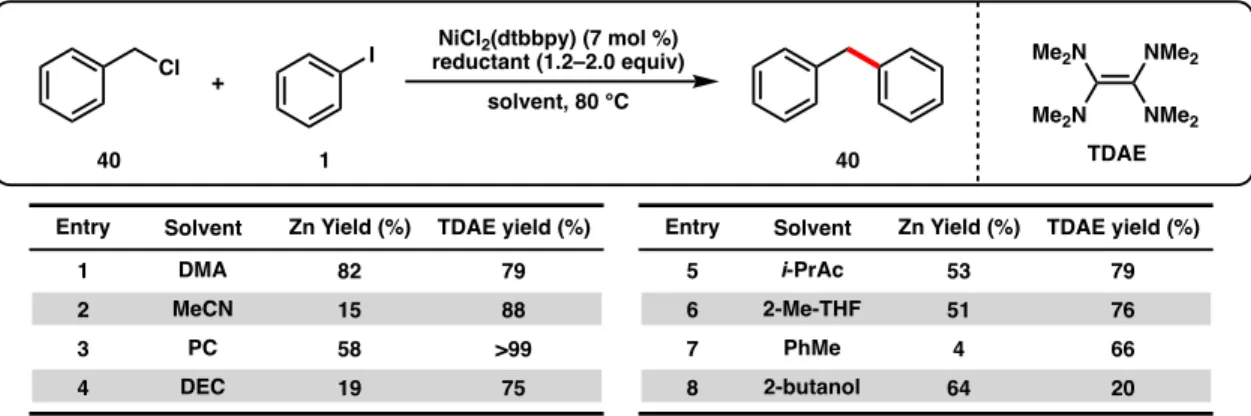
Initial developments in Ni-catalyzed reductive cross-coupling focused on the use of electrochemical approaches using sacrificial metal anodes as the terminal reducing agent. Weix and co-workers were the first to demonstrate Ni-catalyzed cross-electrophilic couplings with unactivated electrophiles,50,53 and since then a multitude of coupling partners have been extensively investigated, predominantly by the Weix and Gong groups.
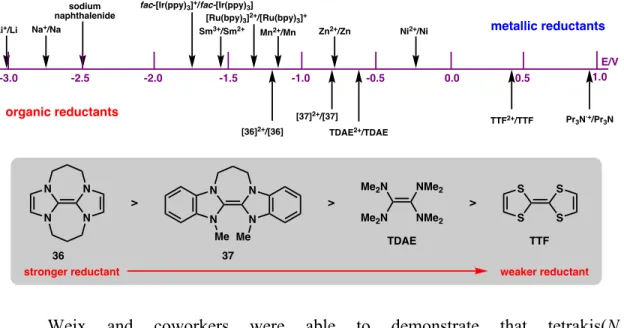
CROSS-COUPLINGS WITH PSEUDOHALIDE ELECTROPHILES
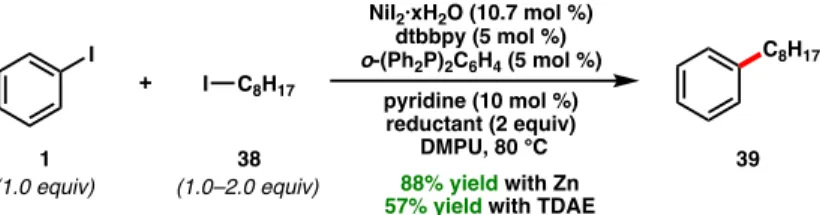
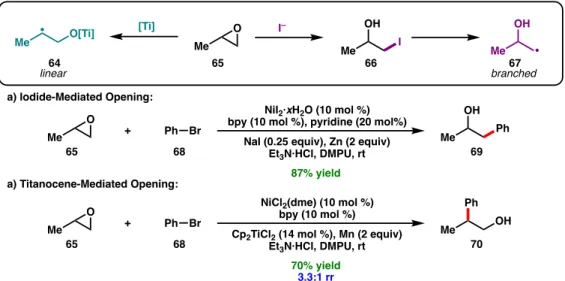
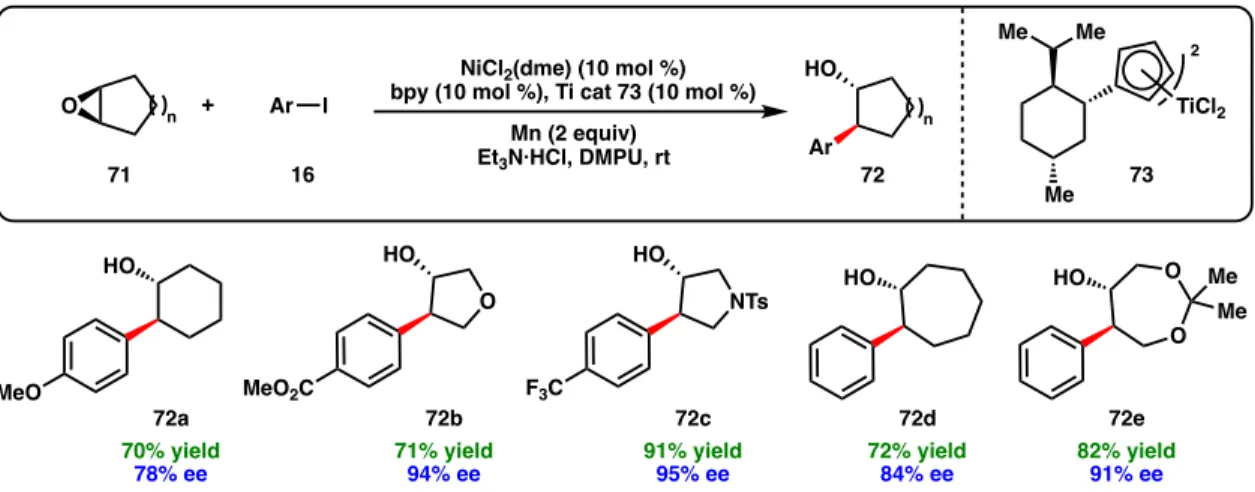
Both methods are compatible; an iterative cross-coupling approach showed chemoselectivity for the aryl bromide over the alkyl bromide to afford 47 (Scheme 1.5c). Weix and co-workers demonstrated that epoxides (65) can be used in regiodivergent reductive cross-couplings to form branched or linear coupling products depending on the mode of epoxide activation (Figure 1.10).
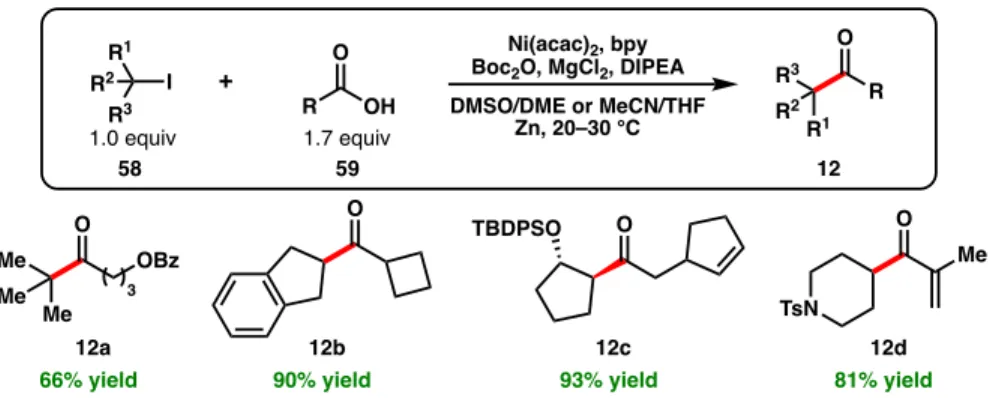
SYNTHETIC APPLICATIONS
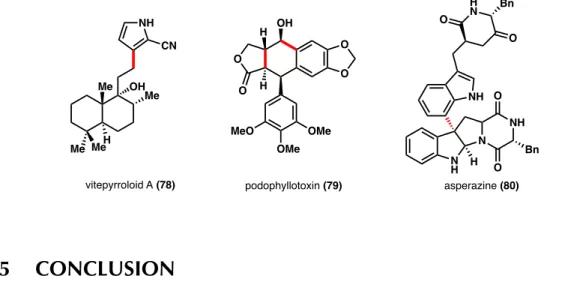
We envision that future advances in Ni-catalyzed cross-electrophilic couplings will continue efforts to intercept feedstock alkenes. Ni-catalyzed reductive cross-couplings have also found recent application in the synthesis of various natural products, including vitepyrroloid A (78), 134 podophyllotoxin (79), 135 and asperazine (80) 136 (Figure 1.13).
CONCLUSION
While asymmetric variants of inter-electrophilic bonds have yet to be applied to natural product synthesis, we anticipate that the continued development of new asymmetric methods will eventually find its use in this synthetic context.
INTRODUCTION
Synthesis and utility of chiral allylic silanes prepared via Ni-catalyzed asymmetric reductive cross-coupling The reaction proceeds with excellent diastereoselectivity and transfer of chirality, yielding the syn-homoallylic alcohol (82) as the major diastereomer. Despite the utility of chiral allylic silanes, the enantioselective preparation of these reagents often requires multi-step sequences or the incorporation of specific functional groups to direct the formation of the C(sp3)-Si bond. Chiral allylic silanes are most commonly prepared through diastereoselective or stereospecific transformations, including Claisen rearrangement of vinyl silanes, bis-silylation of allylic alcohols, silylene insertion of allylic ethers, and the alkenylation of 1,1-silaboronates. ,35 In addition, several enantioselective transition metal-catalyzed reactions have been developed, including the hydrosilylation of dienes,36,37 the silylboration of allenes,38 the introduction of metal carbenoids into Si-H bonds,39,40 and conjugate addition41, 42 and allylic substitution43 reactions.
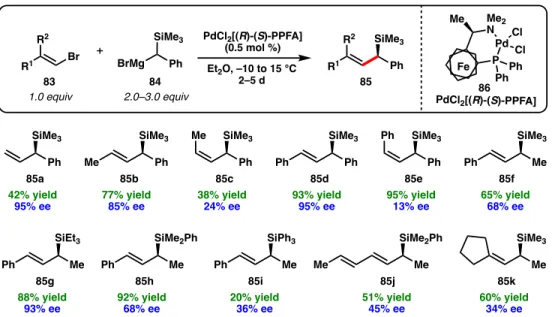
The formation of C-Si bonds is developed in a racemic sense, allowing uncomplicated functional group interconversions (FGI). The ability to access chiral stereocenters via C-Si bond formation remains to be considered.44-49 In contrast, the use of Si-containing nucleophiles or electrophiles in asymmetric transition metal-catalyzed cross-coupling, where the critical silicon-bearing C (sp3)-hybridized stereogenic center is established in the step of forming the C-C bond, represents an alternative and highly modular approach to synthesize chiral allylic silanes (Figure 2.1). The first synthesis of an enantiover-enriched chiral allylic silane was the Pd-catalyzed asymmetric cross-coupling between α-(trimethylsilyl)benzyl magnesium bromide (84) and alkenyl bromides (83), reported by Kumada, Hayashi and colleagues in 1982; These results were followed up by a follow-up study in 1986 (Figure). A variety of alkenyl bromides were evaluated, yielding chiral allylic silane products containing alkyl and phenyl substituents (85a-k). However, this method uses Grignard reagents as coupling partners which are not stable on long-term storage and reduce the functional group compatibility of the reaction.
We envisioned that a Ni-catalyzed asymmetric reductive alkenylation would address the limitation of reagent stability and functional group compatibility, as the required (chlorobenzyl)silanes (34) are bench-stable compounds and reductive cross-coupling reactions typically exhibit good functional group tolerance (Scheme) Thus , a Ni-catalyzed reductive alkenylation can provide chiral allylic silanes not readily accessible by other methods. Herein, we describe a Ni-catalyzed asymmetric reductive cross-coupling to directly prepare enantioenriched allylic silanes (36) from simple building blocks (29 and 34). The resulting chiral allylic silanes are shown to undergo a variety of post-coupling transformations that proceed with high levels of chirality transfer.
REACTION OPTIMIZATION EXPAND .1 Initial Reaction Hit .1 Initial Reaction Hit
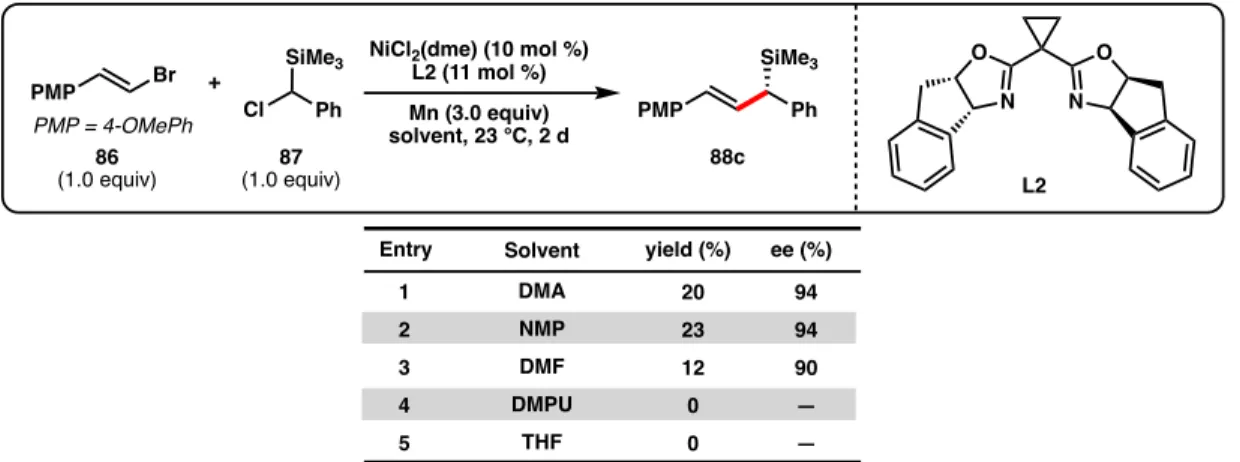
A number of solvents were evaluated in the cross-coupling reaction (Table 2.1) at room temperature (23 °C) in the absence of NaI. A number of other Co sources were also evaluated, many of which simultaneously decreased the ee and reaction yield of 88c (entries 5–10). Overall, it is likely that the co-catalyst phthalocyanine scaffold plays a more important role in improving the reaction yield compared to the identity of the coordinated metal center (Co vs. Fe).
During the course of reaction optimization, we discovered that lowering the temperature improved the reaction yield (Table 2.3, entries 1–3), but achieving full conversion was inconsistent when performed at 0 °C. Therefore, in order to ensure complete conversion, the reaction was performed at 5 °C which consistently provided full conversion for reactions performed in the Freeslate system as well as in a benchtop cryocooling system. The yield of 88c is then maintained between 24–48 h, demonstrating that no decomposition of 88c occurs under the reaction conditions.
In summary, the cross-coupling between 86 and 87 was improved from our previous alkenylation conditions due to: 1) increasing the temperature to 5 °C, 2) running the reaction for 2 days, 3) adding 5 mol % CoPc , and 4) increasing the alkenyl bromide to 2.0 equivalents. A variety of other chiral ligands were evaluated for the cross-coupling between 86 and 87 either at room temperature with 1 mol % CoPc (Figure 2.5) or under the optimized reaction conditions (Figure 2.6). Although chiral BOX ligand L6 provided 88c in excellent enantioselectivity (90% ee) and in higher yield compared to L2 at room temperature, after cooling the reaction to 5 °C, L2 gave a higher yield of 88c compared to L6.
SUBSTRATE SCOPE
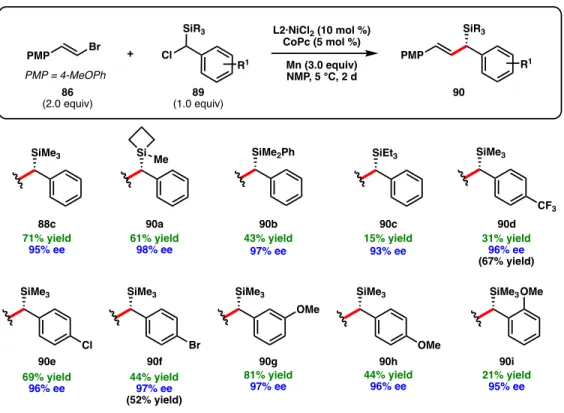
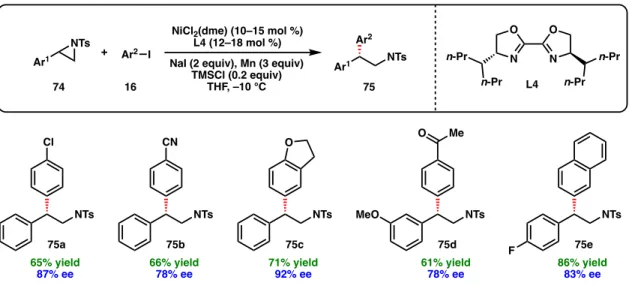
A series of other ligand scaffolds were evaluated at 5 °C, and the desired allylic silane was also formed in excellent enantioselectivity when using chiral BOX ligand L6, BiOx ligand L4 and CyanoBOX ligand L17 respectively and –89% ee) (Figure 2.6). For non-polar alkenyl bromide substrates, m-methoxy chlorobenzyl silane 89g was used as the coupling partner to facilitate product purification (91o–91u). Finally, alkenyl bromides bearing furan (91q), thiophene (91r), pyridine (91k), pyrimidine (91l), and indole (91e) heterocycles can be used, giving the corresponding allylic silanes in high ee.
For example, Z-alkenes (92) and tri-(94, 95) and tetra-substituted (93) alkenyl bromides failed to react to produce the desired coupling products. The use of chlorobenzylsilane containing a free Si-OH group did not yield the desired product. Isolated yields are provided; ee is determined by SFC or HPLC using a chiral stationary phase.
A slight increase in temperature helped to increase the conversion of 104 compared to the reaction run at 5 °C. However, the starting material still remained in the reaction after 2 days, contributing to the lower yield of 88c (Scheme 2.3a). Enol triflate 105 also underwent cross-coupling to give 91a in 57% yield, again with excellent enantioselectivity (Scheme 2.3b).
UTILITY OF CHIRAL ALLYLIC SILANES
Gouverneur and co-workers demonstrated that allylic products CF3 (108) can be synthesized from chiral allylic silanes (107) by photoredox catalysis with Ru(bpy)3Cl2 in the presence of Togni's reagent (Scheme 2.5a).61 The reaction is thought to proceed through the addition of a stereospecific of the trifluoromethyl radical to the alkene, followed by elimination of the trimethylsilane. Hayashi and co-workers demonstrated that allylic silanes (109) can undergo stereospecific protodesilylation with deuterated acetic acid to form 110 with complete enantiospecificity (Scheme 2.5b).62 In a separate report, Hayashi and co-workers showed that epoxidation of the alkene in 91p with mCPBA gives a chiral epoxide that can undergo acid-catalyzed ring opening to give a chiral allylic alcohol with only slight Scheme 2.6 Stereochemical result of epoxidation of a chiral allylic silane. The E and Z isomers are formed in opposite enantiomeric series due to the counter attack of mCPBA on the major and minor conformations of 112 during the epoxidation reaction (Scheme 2.6), which ultimately requires resolution if oxidative cleavage of the alkene is planned.
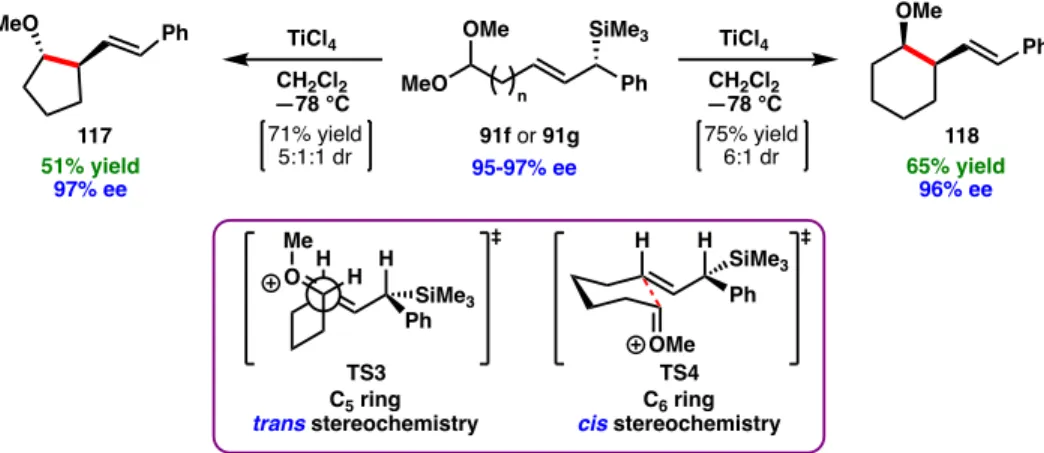
Finally, ozonolysis of the styrene and reduction of the amide (+)-tashiromine (122) provided in 68% yield over two steps. The selectivity of the acid-catalyzed cyclization of 120 was found to be dependent on the identity of the acid (Table 2.8). The use of neat formic acid (HCO2H) tested the best ratio of all acids; 121 was obtained as 68% of the total mixture.
Yields are reported for the combined mixture of isomers, and the major isomer as depicted is given as a percentage of the total mixture (representative purity). Excellent diastereoselectivity and E:Z ratio of the alkene group were observed in most cases, with the exception of reduced dr for 123b, 123e, 124d and 124e. Interestingly, tetrahydrofuran 123e was formed preferentially via SN2 cyclization to yield the 5-membered heterocycle rather than the 6-membered heterocycle.
CONCLUSION
EXPERIMENTAL SECTION .1 Materials and Methods .1 Materials and Methods
The solution was concentrated under reduced pressure and analyzed by 1 H NMR to obtain the reaction yield. The reaction was slowly added to a solution of NaHCO 3 , extracted with Et 2 O, dried with Na 2 SO 4 , filtered and concentrated under reduced pressure. The reaction was quenched with aqueous NaHCO 3 , extracted with Et 2 O, dried with Na 2 SO 4 , filtered over a plug of silica with 50% Et 2 O/hexane and concentrated under reduced pressure to give the crude material.
The reaction was quenched with aqueous NaHCO3, extracted with Et2O, dried over MgSO4, filtered and concentrated under reduced pressure. The reaction was removed from the stir plate and slowly added to a strong stirring solution of Et2O (150 mL) and hexane (150 mL), filtered and concentrated under reduced pressure. The reaction was quenched with aqueous NH 4 Cl (3 mL), diluted with pentane (12 mL) and the layers were separated.
The reaction was warmed to room temperature and stirred for 2 h, then concentrated under reduced pressure. The reaction was diluted (5 mL water + 5 mL Et2O) and stirred while quenching with slow addition of 1 M HCl (2 mL). The reaction was stirred at 0 °C and warmed to room temperature over 3 hours.
Then 5% aqueous NaOH (0.6 mL) was added and the reaction was stirred at room temperature for 8 hours. The crude material was purified by preparative TLC (silica, 20% EtOAc/hexane) to afford 5.7 mg of S26 (57% yield over 2 steps) as a colorless oil. 100 equiv) and methyl iodide (7 µl, 0.105 mmol, 3.5 equiv) were then added, and the reaction was stirred at room temperature for 30 min.
![New 6-(3-Indolyl)benzo[b]carbazoles](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)