The first two people I want to thank are Theodor Agapie and Nilay Hazari. I would like to thank Sara Klamon, who is one of the strongest people I know and yet an extremely fun person to be around.
Two possible outer-sphere C-H bond
The proposed mechanism for the functionalization of
The oxidation of methane to methyl bisulfate reported by Periana et al. and catalyzed by (bipyrimidine)dichloro-
The proposed mechanism for bipyrimidineplatinum(II)-
A potential catalytic cycle for oxidative iridium-catalyzed
A number of enzymes have been shown to efficiently and selectively catalyze the oxidation of alkanes, and these enzymes operate at physiological temperatures and pressures that are mild compared to the conditions required for the heterogeneous conversion of alkanes.4,5,8,9. An additional advantage of enzyme-mediated functionalization of alkanes is that it involves the use of degradable catalysts and non-toxic cosubstrates.9 Unfortunately, large-scale catalysis is currently impractical due to low enzyme activity and stability and the demand for expensive electron donors in the form of cofactors.9.
Two possible outer-sphere C-H bond functionalization pathways
This thesis focuses on determining the requirements for selective, homogeneous, inner-sphere C-H bond functionalization using complexes of platinum and iridium.
Inner-sphere C-H bond functionalization
CuCl2 was shown to catalyze the oxidation of 1 by dioxygen; However, [PtIICl4]2- was also oxidized under these conditions. Increasing the chloride reduces the kox/kH+ ratio by 1 when Na2[PtIVCl6] is used as the oxidizing agent, but when CuCl2 is used as the oxidizing agent, the added chloride increases the kox/kH+.
The proposed mechanism for the functionalization of alkanes catalyzed by platinum(II/IV) salts. 2
It wouldn't even be affected by CuCl2. the reverse reaction occurs: see above) or only O2. In a separate experiment, methyl chloride was found to decompose under reaction conditions in the absence of Cs2[Pt(CH3)2Cl4], while methane, ethane, and carbon dioxide were stable.) The final volume of solution in the vessel was measured at 368 K with measuring cylinder to determine exact concentrations of reagents.
![Table 2.1. The k ox /k H+ for a variety of oxidants reacting with [Pt II (CH 3 )Cl 3 ] 2- (1) at constant [H + ]](https://thumb-ap.123doks.com/thumbv2/123dok/10401659.0/41.918.347.655.632.932/table-variety-oxidants-reacting-pt-ii-ch-constant.webp)
Possible platinum(II) C-H bond activation routes
The absence of peaks corresponding to the 3- and 3'-protons of 2,2'-biindolyl is probably due to deuteration at these positions; deuteration in the 3-position of indole has previously been demonstrated in acidic solutions.19. At the same rate as the Pt-CH3 peak of 3 appears in the 1H NMR spectrum, new peaks are observed in the aryl region for at least one species, possibly 3, containing 2,2'-biindolyl. Unfortunately, the nature of this species is unclear due to the presence of overlapping multiplets in the aryl region.
New peaks corresponding to CH4, CH3D, and CH2D2 were evident along with new peaks in the aryl region. The absence of peaks corresponding to the 3 and 3' protons of 2,2'-biindolyl is probably due to deuteration at these positions; deuteration at the 3-position of the indole was demonstrated in acidic solutions. Over about 4 days, the 1H NMR peaks corresponding to 2 slowly disappeared, while new biindolyl peaks grew along with CH4, CH3D, and CH2D2.
Over the course of about 4 hours, the 1 H NMR peaks corresponding to 2 slowly disappeared, while peaks corresponding to CH4 and CH3D grew along with a new set of peaks that probably corresponded to a. acetonitrile)methylplatinum(II) complex (3) containing 2,2'-biindolyl.
Potential binding of 2 to iridium(III)
The relatively trans-effective functionality of the imidazolyl carbene should facilitate ligand substitution.7 Iridium(III) complexes often suffer from extremely slow rates of ligand substitution, and this can hinder C-H bond activation, which is likely to require the initial coordination of the C-H bond for metal centers.8-11. Currently, there are very few examples of imidazolyl-carbene diphenolate ligands.28,29,30 A precursor of the imidazolyl-carbene diphenolate ligand (3a) has been synthesized previously, in which a methylene unit connects each phenolate to the imidazolyl ring, as shown in 4. .30 The deprotonated ligand is unstable to a 1,2 benzyl migration reaction at room temperature; So the metallization must be done at low temperature. Published reports on the synthesis and metalation of a diphenolate imidazolyl-carbene ligand such as 2 (Scheme 4.3), in which the methylenes do not link the phenolates to the imidazolyl ring, are lacking; although, the synthesis of a similar ligand for ruthenium was reported concurrently with the work described below.31 This type of ligand is predicted to be more thermally robust and form more stable six-membered metallacycles.
The synthesis of titanium and zirconium complexes with a diphenolate imidazolyl-carbene ligand as demonstrated by Kawaguchi et al.30. The precursor of the diphenolate imidazole carbene ligand, 1,3-di(2-hydroxy-5-tert-butylphenyl)imidazolium chloride (2a), was synthesized in 42% overall yield using the procedure illustrated in Scheme 4.5 . This procedure was adapted from the synthesis of monophenolate imidazolium salts by Waltman et al.32 Another potential precursor for an imidazole carbene ligand, 1,3-di(2-bromo-4-tert-butylphenyl)imidazolium chloride (4a) is synthesized in a similar manner (Scheme 4.5).
It was predicted that activation of the bromo-phenyl bonds of 4a with iridium could generate an iridium complex with a tridentate diphenyl imidazolyl carbene ligand.
The synthesis of potential ligand precursors, 2a and 4a. Overall yields are shown
Metallation of 2a to generate 6
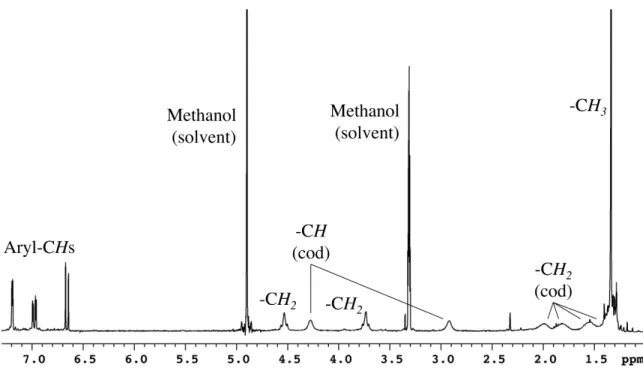
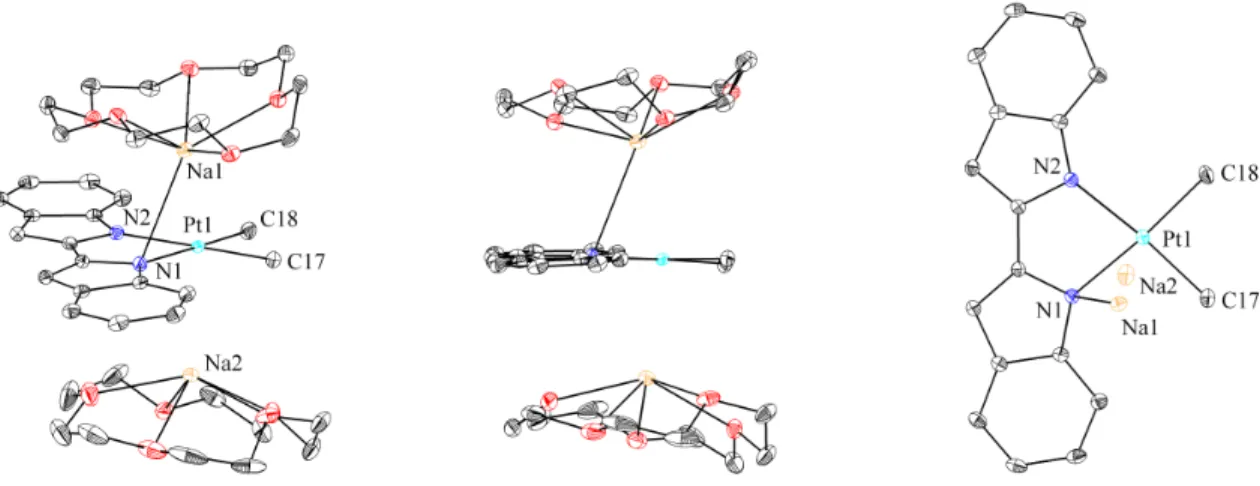
A group of peaks is observed in 1H NMR (fig. 4.2) for both phenolates, and only three peaks are observed for cyclooctadiene. Unfortunately, it is still not clear why two peaks are observed in the 1H NMR spectra for the imidazolyl-CH2 groups. In both C6D6 and high concentrations in THF-d8, the 1H NMR peaks for 6 in the absence of the 18-crown-6 ether broaden significantly.
In C6D6 this interaction can be disrupted in the presence of 18-crown-6 ether as indicated by the sharpening of the 1H NMR peaks for 6. The rest of the peaks are quite sharp with one set of peaks for the phenolates and two imidazolyl – CH2 peaks. It is unclear why in methanol-d4 the 1H NMR peaks for coordinated cyclooctadiene split into two sets.
In the 1H NMR spectrum, there is a cluster of peaks for the two phenolates; there are two peaks for the –CH2 groups of the imidazolyl ring; and, there are six peaks for the iridium-coordinated cyclooctadiene.
The synthesis of 9 and 10 from 6
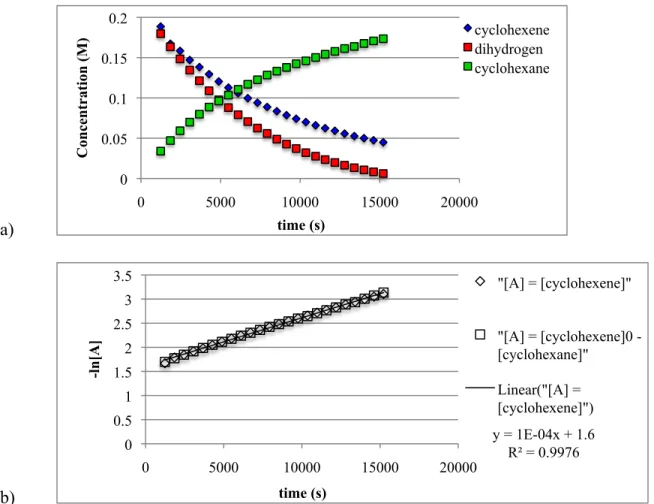
This seems to indicate a zero-order dependence on dihydrogen concentration, meaning that the reaction of the catalyst with dihydrogen is much faster than the reaction of the catalyst with cyclohexene; however, further studies need to be performed to confirm this preliminary result. one). By 1H NMR spectroscopy, only trace amounts of cyclooctene were ever observed; no iridium-hydride signals were observed; and only broad peaks were observed for the diphenolate carbene ligand. Furthermore, it would be difficult to observe an iridium hydride because of the broadening that occurs to the peaks of the other ligands bound to the iridium.
To further explore the coordination environment of the iridium(III) carbene diphenolate complexes, 9 was reacted in separate experiments with trimethylphosphine and tricyclohexylphosphine as shown in Scheme 4.10. Upon reaction with trimethylphosphine, three equivalents of phosphine reacted to form tris(trimethylphosphine){1,3-di(2-hydroxy-5-tert-butylphenyl)imidazolyl}iridium(III) hexafluorophosphate (11) as shown in Scheme 4.10; . Regarding 12, only one singlet for the coordinated tricyclohexylphosphines is observed in the 31P NMR spectrum and only one set of peaks in the 1H NMR spectrum for both phenolates.
The 1 H NMR spectrum of 11 also contains only one set of peaks for both phenolates, but the 31 P NMR spectrum of 11 contains a doublet and a triplet with an intensity ratio of 2:1.
Displacements of cyclooctadiene from 9 with trialkylphosphines
The reaction of 12 with 1.5 equivalents of trimethylphosphine
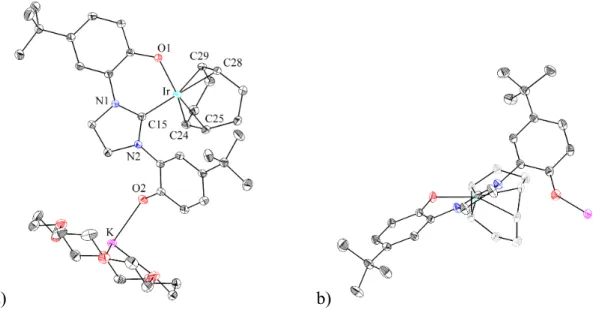
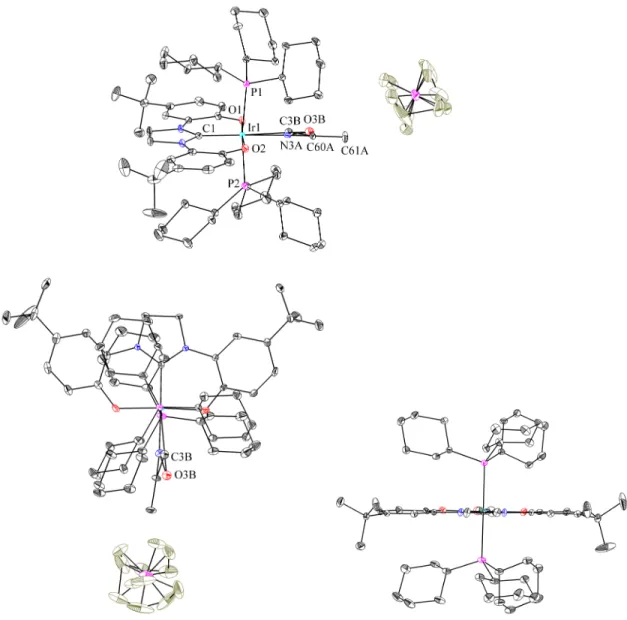
This indicates that significant amounts of bis(tricyclohexylphosphine)(trimethylphosphine){1,3-di(2-hydroxy-5-tert-.butylphenyl)imidazolyl}iridium(III)hexafluorophosphate were not formed, probably due to the application of substantial steric constraints of tricyclohexylphosphine ligands. Determination of an equilibrium constant is complicated by the formation of yellow crystals above the surface of the solution during the reaction. Both complexes have the diphenolate carbene ligand coordinated meridionally with the tricyclohexylphosphine ligands trans to each other as shown in Figure 4.9.
It should be noted that the diphenolate carbene ligand is relatively shallow in this structure with a Cipso-O1-O2-Cipso torsion angle of only 2.9˚. 9 was also reacted with carbon-13C-monoxide to confirm the solution-phase geometry of 15 and to check for an isotope shift in the infrared spectrum. The resulting iridium(III) complex containing carbon-13C-monoxide (15') exhibits a triplet in the 13C NMR spectrum and a doublet in the 31P NMR spectrum as expected.
Equilibrium between 12 and 15 which occurs in acetonitrile at 90 ˚C under an atmosphere of carbon monoxide
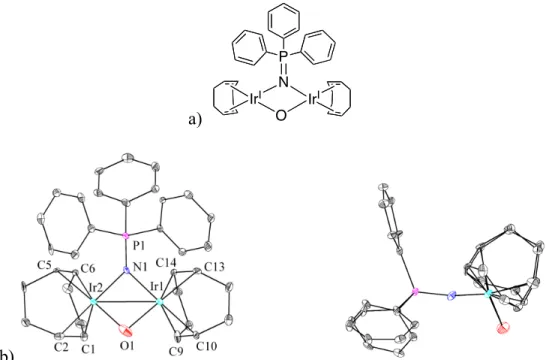
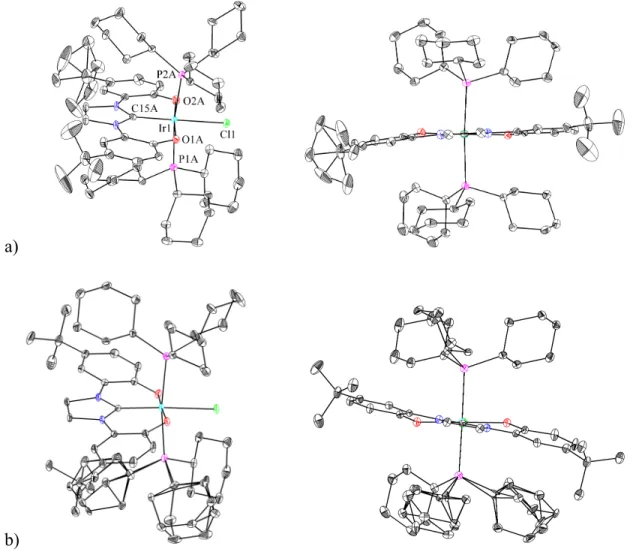
While the crystal structure shown in Figure 4.9 of 15 and 12 could provide additional insight into iridium–carbonyl bonding, this analysis is precluded by inaccuracy in the bond lengths, possibly due to only 22% occupancy of 15 in the unit cell. Crystals of 16 were obtained from methylene chloride (CH2Cl2) and the structure, as determined by single crystal X-ray diffraction, is illustrated in Figure 4.10. Two isomers of 16 (16' and 16'') occupy the unit cell together with two molecules of methylene chloride.
The metal center, the chloride, the two oxygens and the carbene carbon lie approximately in one plane for. To compare the degree to which the diphenolatimidazolyl ligand of each isomer is distorted from C2v symmetry to C2 symmetry, Cipso-O1-O2-Cipso. Thus, the diphenolatimidazolyl ligand of 16'' is significantly more distorted from C2v symmetry than the corresponding ligand of 16'.
However, it should be noted that there is some disorder in the crystal structure, and this could affect the observed angles; moreover, differences between the two isomers may be caused to some extent by the packing of the molecules in the crystal.
Reaction of 12 with tetramethylammonium chloride to generate 16
Coulometric oxidation of 16 at a positive potential of the first oxidation wave (0.25 V) resulted in the passage of 0.92 faradays per mole, indicating that the wave at -0.22 V corresponds to a one-electron oxidation of 16 .EPR mass spectroscopy of the sample from controlled potential electrolysis showed a molecular ion with mass 16. EPR spectrum at 7 K of the solution resulting from a coulometric oxidation (0.92 faraday per mole) of 16.
Mass spectrometry of the EPR sample obtained from the second coulometric oxidation of 16 indicated a molecular ion with the mass of 16 . EPR spectrum at 18 K of the solution resulting from two coulometric oxidations (each about 1 faraday per mol) of 16. Depiction of the HOMO for the fully optimized gas-phase structure of 16 determined by QMMM calculations.
This interaction appears to cause a distortion of the diphenolate-imidazolyl carbene ligand from C2v symmetry to C2 symmetry as observed in the solid state structure of 16. Coulometric oxidation of 16 was performed at a potential positive of the first oxidation wave (0.25) V ), and the results are shown in figure 4.23. Coulometric oxidation of 16 was continued at a potential positive of the second oxidation wave (0.96 V), and the results are shown in figure 4.24.
![Figure 2.2. The dependence of k MeCl /k MeH for 1 on [Cu 2+ ] (left) and [Fe 3+ ] (right) ([H + ]=](https://thumb-ap.123doks.com/thumbv2/123dok/10401659.0/42.918.175.799.514.796/figure-dependence-mecl-meh-cu-left-fe-right.webp)
![Figure 2.1. The dependence of k MeH /k MeCl for 1 on [H + ] ([Cu 2+ ] = 0.05 M, [Cl - ] = 4 M).](https://thumb-ap.123doks.com/thumbv2/123dok/10401659.0/42.918.167.455.128.397/figure-dependence-meh-mecl-h-cu-05-cl.webp)