Mingjie Jin, who shaped me to be the person I am and who were the role models for the person I aspire to be. Furthermore, a general computational scheme is presented that also allows for the rapid enantioselectivity prediction of many related pybox ligands, to help predict the next generation of asymmetric Negishi coupling catalysts.
Iridium and Rhodium analogues of the Shilov cycle cat- alystalyst
Much of the focus has been on pathways involving C–H activation in a transition metal center followed by oxidative cleavage of the resulting C–M bond, which offer greater potential for good selectivity by avoiding radical-based pathways. In this chapter, a computational screening of a variety of ligands was performed to find a Rh-ligand complex with the predicted optimal combination of low methane activation energy, low methyl group functionalization energy and overoxidation protection of the MeX product.
Investigation and applications of the Reduction-Coupled Oxo Activation (ROA) mechanistic motif towards alkaneOxo Activation (ROA) mechanistic motif towards alkane
The stability of the resulting mono-hydrogenated species has been attributed to the fact that spin density appears to be localized on the neighboring vanadium atoms (Figure 1.1). Specifically, questions such as the generalizability of the mechanism to other metal-headgroup pairs, as well as the applications of the ROA motif to homogeneous catalysts, are addressed.
Other projects
This means that the first step is endothermic by only 5.0 kcal/mol, with a later calculated activation energy of 13.6 kcal/mol, which is within the experimental range. We designed a homogeneous complex κ-P bis(2-phenoxyl)phosphinite vanadium ((OPO)V) as a catalyst capable of activating propane using the ROA mechanistic motif.
Abstract
Introduction
Results and Discussion
Synthesis and Characterization of Ir I complexes
The closest Na (15-crown-5) lies above the plane of the ligand and is omitted for clarity. The synthesis of the ligand, which was straightforward and analogous to that of H2(ONO), is shown in Scheme 2.2.
Synthesis and Characterization of Ir III Complexes
The crystal structure of 8 is shown in Figure 2.7. 2.1) Exposure of 8 to water followed by proton sponge addition at room temperature gave trans-(ONOtBu)Ir(PPh3)2OH (9, eq 2.2). The proposed mechanism for this transformation is single-electron transfer followed by H atom abstraction (Scheme 2.5); the shown byproduct of oxidized proton sponge was observed (quantitatively).
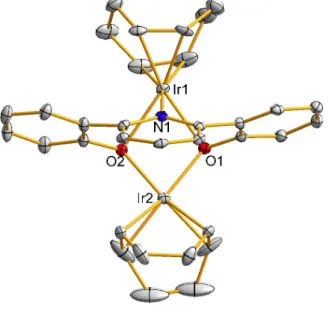
C−H Activation by Ir III (ONO tBu ) Complexes
The reaction of 14 with AgPF6 gave dark blue [(ONOtBu)Ir(PEt3)2Me]PF6 (16, Scheme 2.7), characterized by a crystal structure and an EPR spectrum (Figure 2.15) very similar to those of (relatively) few) organometallic IrIV species reported [13]. 1H NMR signals would be expected for the (CH2CN)-methylene protons, which are diastereotopic in the solid state structure, only one is observed; this may be due to either accidental degeneration or dissociation of the dimeric structure in solution.
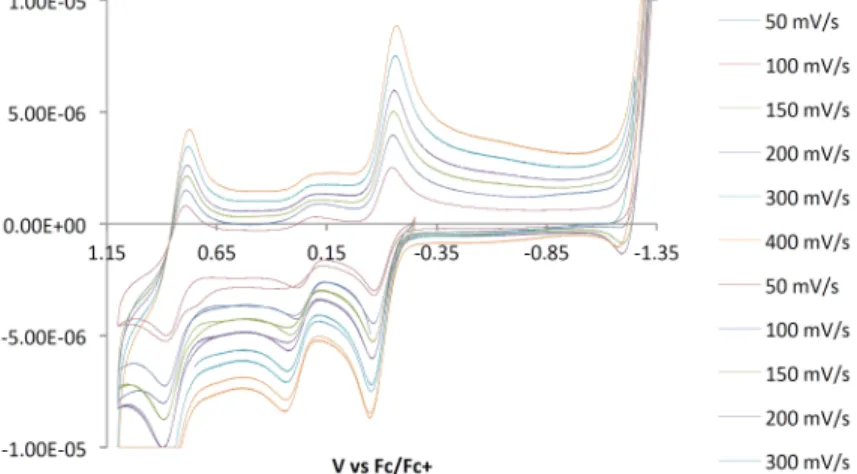
Kinetics of C−H Activation
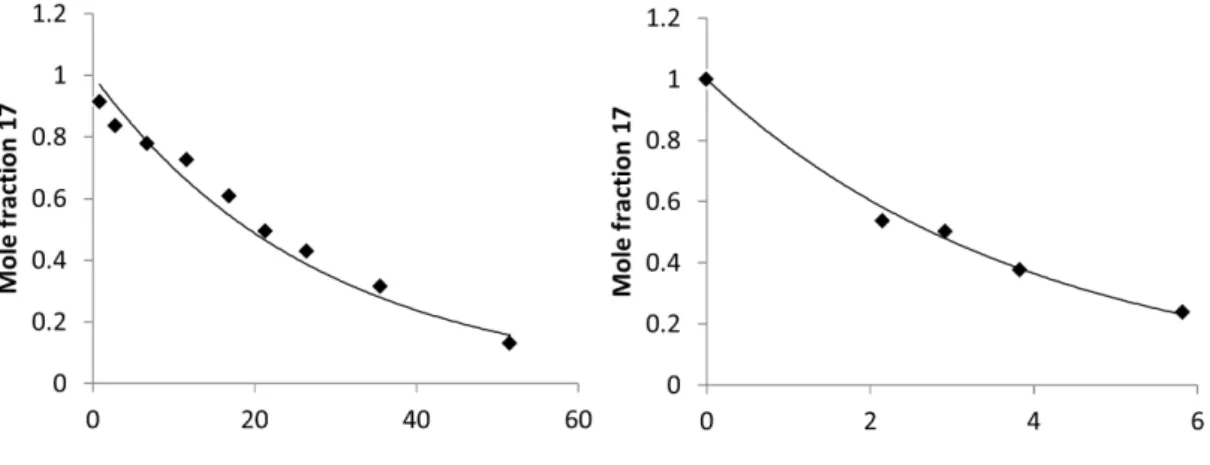
The rate law for direct conversion of 12 to 18 in p-xylene should be identical to the above, and the reaction exhibits pure first-order kinetics with a rate (kobs s−1) about a factor of 2 slower than that of 12 to 17 . In benzene solution, the reaction must be first order17 and independent of [L]; both were observed.
![Figure 2.20. Left: kinetics of conversion of 12 to 17 in C 6 D 6 at 65 ◦ C. Right: kinetics of conversion of 12-d 30 ((ONO tBu )Ir[P(C 6 D 5 ) 3 ] 2 Me) to 17-d 36 ((ONO tBu )Ir[P(C 6 D 5 ) 3 ] 2 C 6 D 5 ) in C 6 D 6 at 65 ◦ C.](https://thumb-ap.123doks.com/thumbv2/123dok/10406696.0/50.892.187.794.333.525/figure-left-kinetics-conversion-right-kinetics-conversion-ono.webp)
Conclusions
For the reaction in p-xylene the starting point is [17] = the maximum [PhH] = 8.8 mM and the minimum value of k5/k4K3[PhH] = 160, thus the assumption necessary to take the kinetics into account of the first order is valid as Good.
Supporting Details
Experimental Section
15-Crown-5 (21 µL, 106 µmol) was mixed and the solution was allowed to stand undisturbed for several days. After two hours, the solution was removed in vacuo, redissolved in pentane and filtered, and the solvent was again removed in vacuo.
Kinetics Details
The specific flow is detailed in the relevant section of Figure 2.23; b) added equivalents of PPh3;. For PPh3 inhibition experiments, the Ir complex was mixed with various amounts of a 20 mg/mL stock solution of PPh3 in C6D6 and additional C6D6 was added to increase the volume to 700 µL.
Crystallographic Data
Acknowledgments
14] The crystal structure included a complex solvent region that could not be satisfactorily modeled, and also showed the presence of Li+ ions, probably carried over as impurities from the previous step throughout the synthetic sequence; the necessary counteranions to balance the charge could not be found, probably because they lie in the solvent region.
Abstract
Introduction
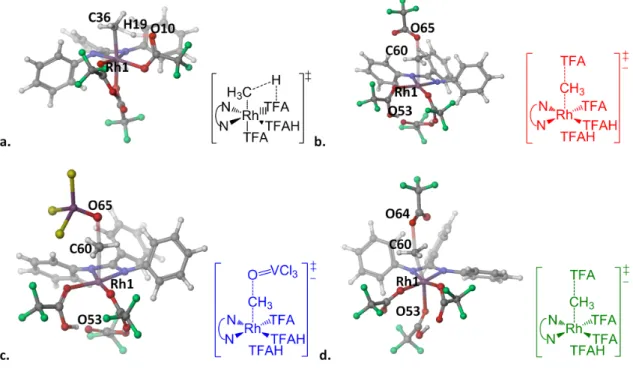
The first case, RhIII(NNF), leads to a transition state barrier of ∆G‡ = 27.6 kcal/mol at 298 K for the activation of methane to TFAH (35.0 kcal/mol in water), the most low that we found using RhIII. The second case, RhIII(bisq), leads to a transition state barrier of ∆G‡ = 33.4 kcal/mol at 298 K for the activation of methane to TFAH, and ∆G‡ = 32.0 kcal/mol at 298 K for Rh−Me functionalization in TFAH, also among the lowest we have found using RhIII.
Materials and Methods
Thus, the free energy of such gas molecules can be simply calculated using the formula. For pure liquids (e.g., trifluoroacetic acid and water), the Gibbs free energy was calculated using the formula
Results
The Rh(NN) complexes in TFAH
Indeed, the activation barrier for this is too high at 54.8 kcal/mol at room temperature (Figure 3.2b). Oxidation from RhIII to RhIV was effective in lowering the global SN2 barrier from 54.8 to 42.2 kcal/mol.
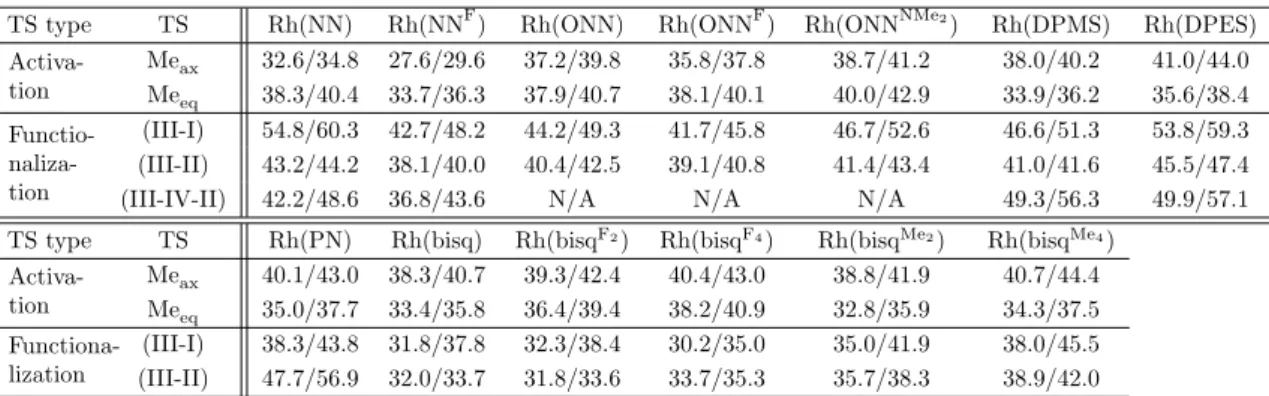
The Rh(NN F ) complexes in TFAH
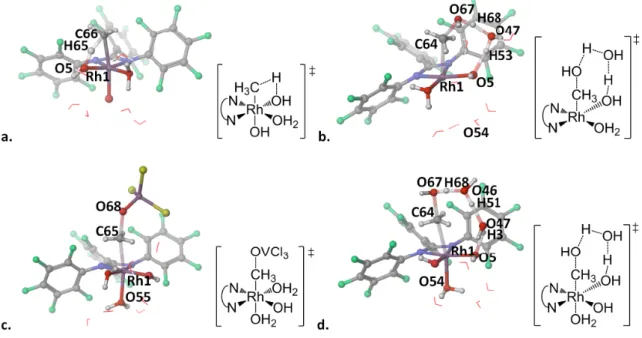
The Rh(NN F ) complexes in water
For the III-II route (Scheme 3.7), we no longer needed to require the leaving group transferred to the RhIII methyl to be a water ligand. For the III-IV-II route (Scheme 3.8), a good leaving group trans to RhIII-methyl again became important.
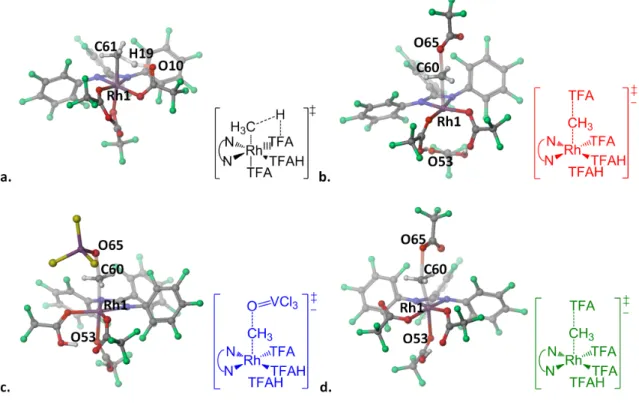
The Rh(ONN x ) family of complexes in TFAH
Catalytic cycle for the activation and functionalization of methane using Rh(ONNF) complexes in TFAH. Catalytic cycle for the activation and functionalization of methane using Rh(ONNNMe2) complexes in TFAH.
The Rh(DPMS) and Rh(DPES) complexes in TFAH
The Rh(PN) complexes in TFAH
Methane activation and functionalization using Rh(DPMS) complexes in TFAH, via the Rh(III-I), Rh(III-II), and Rh(III-IV-II) pathways. Methane activation and functionalization using Rh(DPES) complexes in TFAH, via the Rh(III-I), Rh(III-II), and Rh(III-IV-II) pathways.
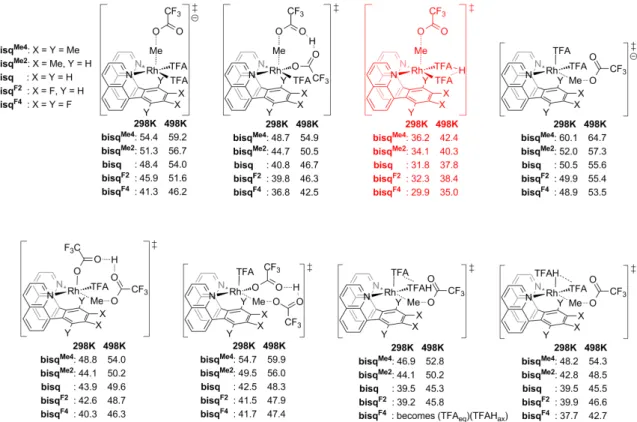
The Rh(bisq x ) family of complexes in TFAH
For the III-I pathway, SN2 attack by TFA–/TFAH results in the formation of the Me-TFA product and RhI(bisqx)(TFA)(TFAH), which is reoxidized to the starting complex RhIII(bisqx) (TFA). 3. Bottom: plot of the activation and III-II functionalization transition state energies of the different Rh(bisqx) complexes.
Product protection and C−H activation
All free energies are in kcal/mol and relative to the lowest energy inorganic state (i.e. just before methane activation). Overall, the product protection is comparable to the Rh(NNx) family at 298 K, but is significantly improved by 3.8–4.2 kcal/mol at 498 K.

Discussion
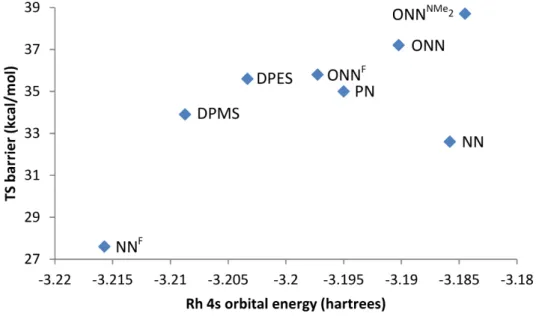
Comparison of various Rh parameters with transition state barriers associated with rhodium-ligand complexes. Free energy of the direct precursor before the lowest activation barrier of methane, in kcal/mol.
Conclusions
The next logical step in our research is the experimental synthesis and stability study of RhIII(NNF) and RhIII(bisq) complexes. In particular, for RhIII complexes with neutral methane activation transition states, increased electron-withdrawing ligands result in lower barriers for activation and functionalization.
Acknowledgments
We can estimate the overall transition state barrier by simply calculating the RhIII−Me species and extracting the Rh−C binding energies. For the RhIII(bisqx) family of complexes, the requirement that one coordination site be a weak η2-benzene interaction greatly facilitates functionalization on a methyl group transitioning thereto.
Abstract
Introduction
The VPO catalyst for n-butane oxidation
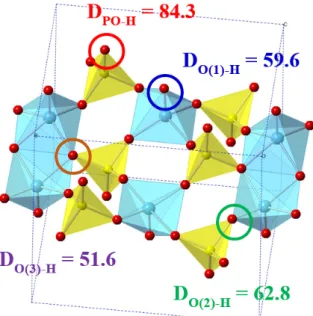
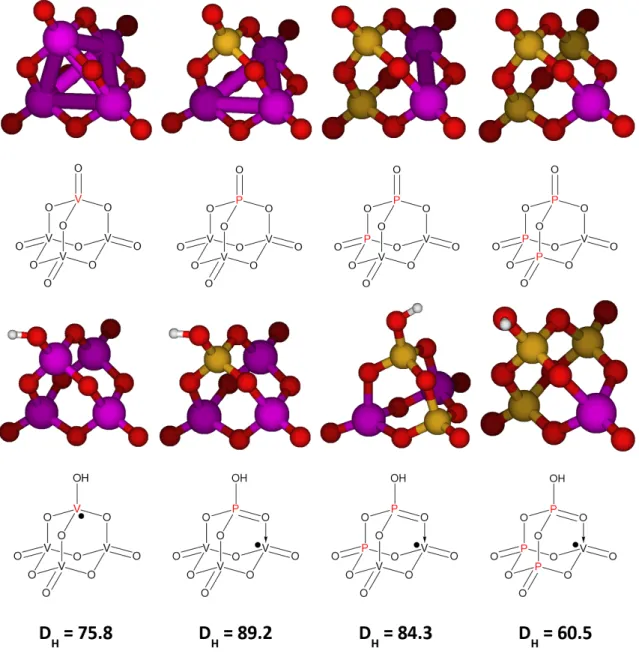
Unfortunately, the DH values calculated for the resulting vanadium peroxo and vanadium oxo moieties were only 70.0 and 58.9 kcal/mol, respectively. Surprisingly, phosphorus oxo was found to have a much higher DH value of 84.3 kcal/mol.
The ROA mechanistic motif
The O-H bond strength of 84.3 kcal/mol with the P-O group leads to a first step that is only 5.0 kcal/mol endothermic, with an activation energy later calculated to be 13.6 kcal/mol. Compared to a pure vanadium (V) oxide shell, coupling to one or two P atoms makes it 13.4 and 8.5 kcal/mol more active, respectively.
Potential homogeneous catalysts utilizing the ROA principle
We therefore propose, in the second part of this chapter, that the hypothetical complex diaqua[bis(2-phenoxy)phosphinite-κ−P]oxidovanadium (V) ((OPO)VVO(H2O)2) uses the reduction-coupled oxo activation (ROA ) mechanistic motif as a catalyst for the oxidation of alkanes in air. We describe the most plausible transition state barriers for the conversion of propane and dioxygen to oxidized products such as propylene and isopropanol.
Materials and methods
6-31G** basis set [21] for the other atoms; while the larger basis set consists of the triple-ζ Los Alamos basis set and pseudopotential (LACV3P**++) for transition metals, the same LAV3P basis set and pseudopotential for heavy main group elements, and the 6-311G**+ + basis set [21] for the other atoms. The free energy for each molecular species in solution was calculated using the formula G=Egas+ ∆Gsolv+ ZPE +Hvib+ 6kT −T[Svib+γ(Strans+Srot−α) +β].
Validation of ROA on small oxide clusters
Optimized structures of the XV3O10 species (first row) (X∈ {P,As,Sb,Bi}and their mono-hydrogenated XV3O10H counterparts (second row). Optimized structures of the Z2V2O11 species (first row) (Z∈ {S, Se,Te}) and their monohydrogenated Z2V2O11H counterparts (second row).
Reactions of a homogeneous oxidation catalyst utilizing the ROA mechanistic motifthe ROA mechanistic motif
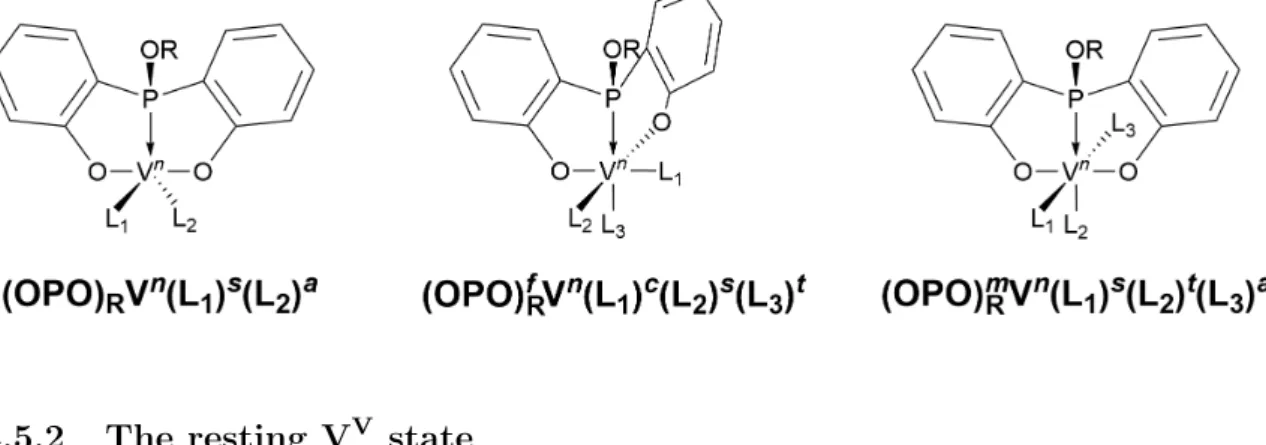
- Nomenclature
- The resting V V state
- Propane activation transition states
- Monohydrogenated V IV states
- Catalytic cycle involving H-atom abstraction only
- Reduced V III states
- Catalytic cycle involving H-atom abstraction followed by isopropyl addition
- Comparison of the catalytic cycles
At pH 0, this species is 30.3 kcal/mol above the neutral ground state (OPO)fHVVOc(OH)s(H2O)t, and with. At pH 0 the lowest energy species is (OPO)fHVIVOc(H2O)st2 at 13.4 kcal/mol above the ground state.
Conclusions
17] The proof radius is automatically calculated by Jaguar using the formula = 0.5833p3 MW/ρ where MW is the molecular weight and ρ is the density of the solvent.
Abstract
Introduction
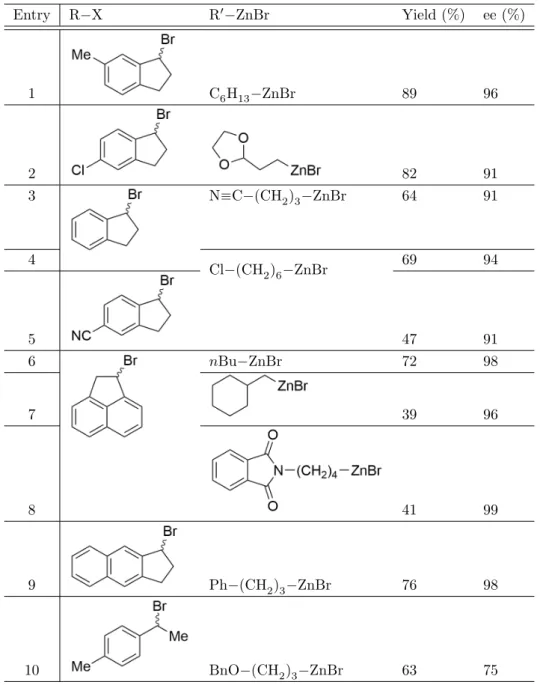
The Ni(iPr−pybox) system
The Ni(iPr−pybox) system, developed by Gregory Fu and co-workers, is a Negishi coupling catalyst using nickel and the ligand 2,6-bis(4-isopropyl-2-oxazolin-2-yl)pyridine, abbreviated as (iPr -pybox), and combines organic halides with organozinc complexes [9]. However, one of the most interesting properties of the Ni(iPr−pybox) system may be its stereoconvergence.
Existing computational work
While the authors claim to investigate the enantioselectivity of the Ni(iPr−pybox) system, in many reactions the simplified pybox ligand (without the isopropyl groups) was used instead of (iPr)-pybox to simplify the calculations;. Only one radical attack pathway (i.e. that of the indanyl radical axially approaching Ni) was considered.
Materials and methods
Results
- The overall general mechanism
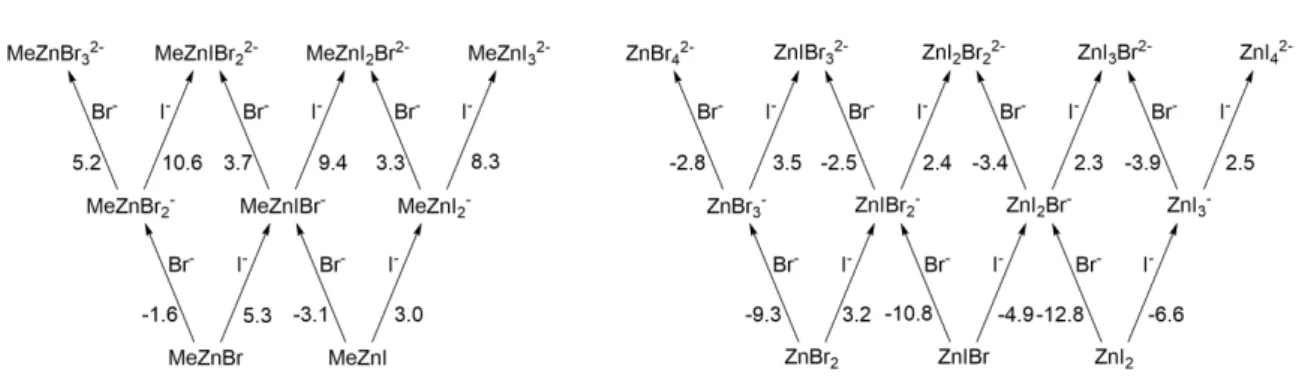
- Nature of the organozinc reagent
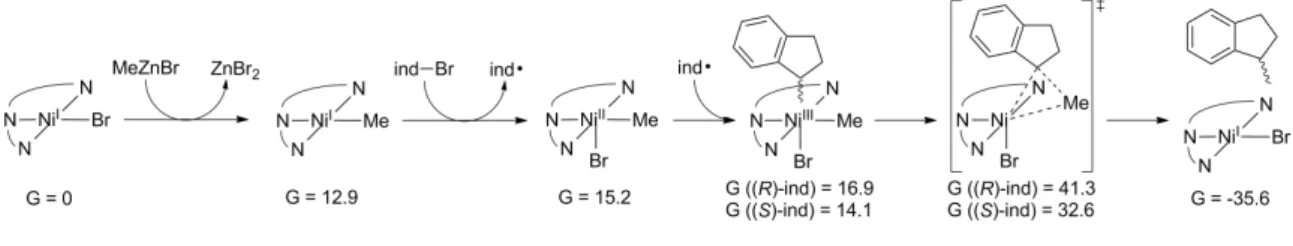
- Mechanism for Ni I oxidation and methylation
- Mechanism for reductive elimination
Methylation of NiI((S, S)−iPr−pybox)Br to NiI((S,S)−iPr−pybox)Me is thermodynamically uphill by 15.2 kcal/mol. Initial coordination of (R)- and (S)-1-bromoindane with NiI((S, S)−iPr−pybox)Br produces an adduct NiI((S, S)−iPr−pybox)(Br)( Br −ind) which are favorable at -4.6 and -5.2 kcal/mol relative to the initial NiIs species, respectively.
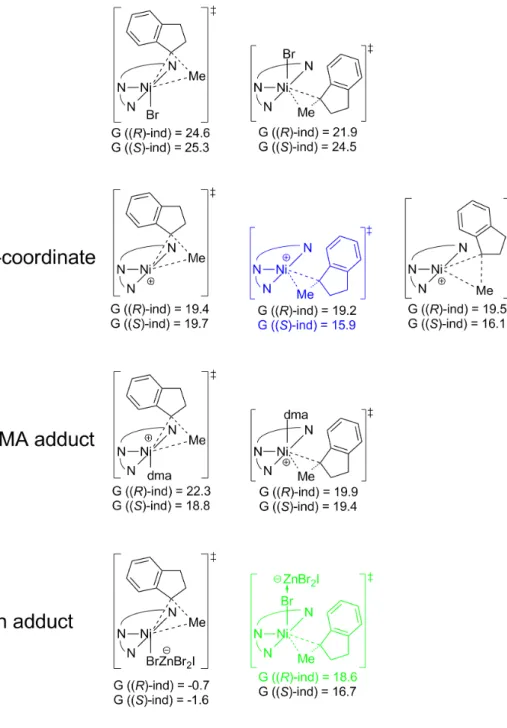
Discussion
Overall catalytic cycle
As a last resort, the only identified transition states containing a Ni−Br−Zn moiety were the geometric isomers [NiIII((S, S)−iPr−pybox)(Me)(ind)−Br−ZnBr2I]–. Therefore, the preference for (S)-1-methylindane is ∆∆G‡ = 2.7 kcal/mol, in agreement with experimentally determined enantiomeric excesses [15].
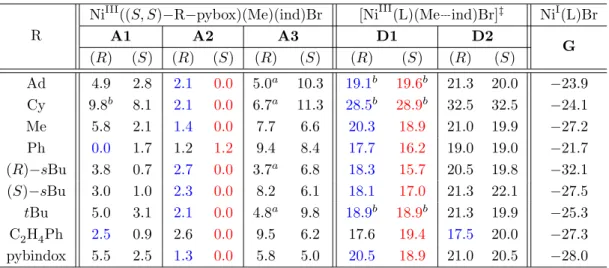
Predictions for new ligands
The lowest transition states for the formation of (R)- and (S)-1-methylindane are marked in green and blue, respectively. The numbers in blue are the lowest energy transition states for the production of (R)-1-methylindane, while the numbers in red are the lowest energy transition states for the production of (S)-1-methylindane.
Conclusions
For each type of transition state, only the energy of the lowest geometrical isomer is reported. Energies given are relative to the lowest calculated species Aneutral NiIII(R−pybox)(Me)(ind)Br complex.
Supporting figures
Numbers in blue are the lowest energy (R)-ind geometric isomers for B and E, while numbers in red are the lowest energy (S)-ind geometric isomers for B and E. b) Geometry optimized for a Meaxindeq (B1/E1) ) configuration. Numbers in blue are the lowest energy (R)-ind geometric isomer for C, while numbers in red are the lowest energy (S)-ind geometric isomer for C.
19] The proof radius is automatically calculated by Jaguar using the formula= 0.5833p3 MW/ρ where MW is the molecular weight and ρ is the density of the solvent.
Abstract
![Figure 2.15. EPR spectrum of [(ONO tBu )Ir(PEt 3 ) 2 Me]PF 6 , 16, in dichloromethane at 20 K.](https://thumb-ap.123doks.com/thumbv2/123dok/10406696.0/42.892.278.693.742.979/figure-epr-spectrum-ono-tbu-ir-pet-dichloromethane.webp)