In fact, some of the theories for molecular electron transfer reactions were developed by analogy with the theories of small polarons in solid state systems. The electronic transmission coefficient (K) controls the rate of electron transfer in the transition state region. If electron transfer is adiabatic, the reactants are confined to the lower surface and move ballistically through the transition state region.
In these studies, it was shown that the rate of electron transfer directly correlates with the longitudinal relaxation time of the solvent medium.
Introduction
The van der Waals energy of each conformation was determined using a Lennard-Jones 6-12 potential function of the shape. 16 Each of the redox ligands prepared in this way was characterized by 1 Hand 31 P NMR spectroscopy. NMR spectra of the ligands show proton chemical shifts and multiplicities for the aromatic protons and ring substituents of pyridinium, which are similar to those observed in spectra of their 1-(2-hydroxyethyl)pyridinium precursors.
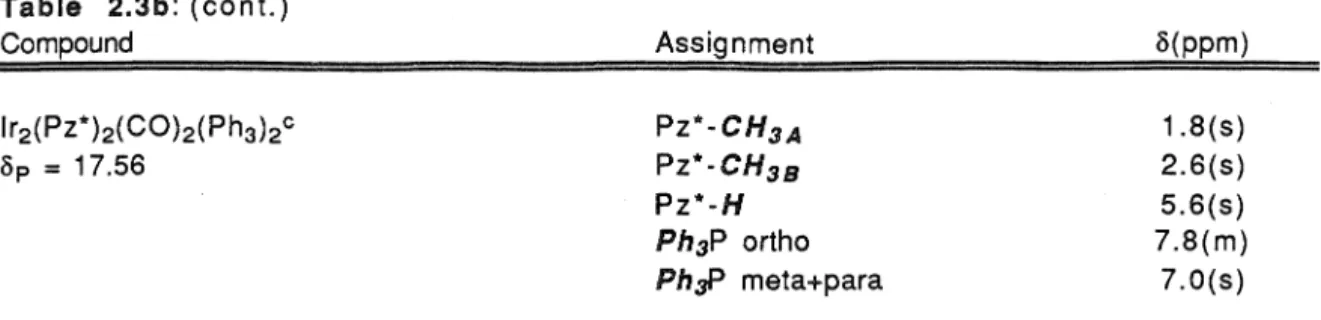
Initial attempts to prepare redoxphosphinite complexes of diiridium bis(µ-pyrazolate) using the two-step method of Stobart and co-workers (Figure 2.4) were complicated by the propensity of the products to form as difficult oils. Additionally, based on the assignments made for the pyrazolate substituents in lr2(Pz*)2(CO)4 (Figure 2.5a), the chemical shifts of the pyrazole methyl groups in lr2(Pz*)2(CO)2{Ph2POCH2CH3 )2 can be assigned according to the labeling scheme seen in figure 2.7. The chiral nature of the complex is manifested in its NMR spectra through the diastereotopic chemical shifts observed for magnetically inequivalent protons bound to the phosphinite ligand alkoxy and phenyl substituents.
The chemical metaproton shifts are not so much affected by the magnetic nonequivalence of the ligand phenyl groups and are observed as a complex multiplet at o = 7.2 ppm. The spectroscopic properties of all the donor-acceptor complexes reported here are similar to those seen in lr2(Pz*)2(C0)2(Ph2POCH2CH3)2 (Table 2.3b). The diastereotopic chemical shifts seen in spectra of the bis-(µ-3,5-dimethyl-4-isobutylpyrazole) complexes indicate that these compounds also have a trans configuration of their terminal ligands.
The familiar duplication of signals for the asymmetric ligand substituents seen earlier is observed at the methyl and ex-methylene protons of the isobutyl groups of the pyrazole bridge (6 = 0.35 and 6 = 1.55 ppm, respectively). 31 P NMR spectra showed a solitary singlet with chemical shifts between 95.02 and 104.29 ppm, consistent with the coordination of phosphinite ligands to the lr(I} metal center). [lr2(Pz*)2(CO)2(Ph2POCH2CH2-Py)2](Ph4B)2 was determined by X-ray crystallographic structure techniques.
These minor deviations may reflect the different steric requirements for the complex's terminal CO and phosphine ligands.

02-C2-IR2 01-Cl-IRl
The bond angles between adjacent ligands in each of the square faces are close to 90°. With the exception of the lr1-C1 and lr2-C2 bonds, the refined structures from both datasets were nearly identical. It was found that the disparity in Ir-CO bond lengths was greater.
Second, structures refined only by high-angle reflections had Ir-CO bond lengths that were the same within the estimated standard deviations. Since low-angle reflections are more sensitive to errors due to heavy atom absorption, this result further confirms that the 0.1 A difference in Ir-CO bond lengths is due to Ir X-ray absorption effects. The bond lengths and angles for the comparable substituents on each of the phosphinite ligands in this complex are the same within their estimated standard.
Furthermore, the torsion angles along the alkoxy substituents of the ligands are similar, indicating that the molecules have implicit C2 symmetry in the solid state. Both ligands adopt a rotational conformation around the lr-P bond, which avoids less favorable steric interactions between the phosphine phenyl groups and the bridging pyrazole ligands. While this solid conformation represents the lower energy configuration of the lr-P rotational coordinate, other conformations are most likely accessible in liquid solution (vida infra).
The Ir-P-O-C and P-O-C-C torsion angles of the ligand alkoxy groups are significantly less than 180°, leaving the three-atom bridging group connecting the pyridinium cation to the metal complex partially extended.

Discussion
Central to the pursuit of this goal is the development of synthetic methodologies that are flexible enough to readily adjust the redox potentials of the donor and acceptor, the electronic properties of the bridging group and the donor-acceptor separation within a range of redox complexes. Studies spanning a range of different inorganic chromophores will be important to understand variations in electron transfer rates as a function of the nature of the excited donor. Structural properties: An understanding of intramolecular electron transfer in the donor-acceptor systems prepared here will be developed by making comparisons between the photophysical properties of the redox-active complexes and their.
The electronic properties of the redox complexes and model compounds are studied in detail in Chapter Three. Rotations about one of its lr-P bonds (A) drastically affect the relative orientation of the two redox partners. Therefore, distances from a pyridinium nitrogen atom to both iridium metal centers are reported with each structure.
Depending on the nature of the interactions between this charge transfer and donor (acceptor) localized states, certain photophysical properties of the donor-acceptor molecules will be disturbed. In the next chapter, the electronic and photophysical properties of the donor-acceptor complexes are investigated using steady-state absorption and emission. spectroscopy and electrochemical techniques. In interpreting spectra from these studies, it was essential to know which of the (dcr*pcr) excited states were involved in electron transfer reactions.
In addition, it was important to characterize the spectroscopic properties of the charge transfer states in these systems. Cuvettes were mounted on a copper block, which was attached to the cold stage of the cryocooler.
Covalent binding of two square planar d8 complexes to a dimeric molecule forces an axial interaction between the d22 and Pz orbitals of the monomeric fragments, leading to the formation of dcr and pcr dimer MOs. Moreover, because their low-energy excited states involve dcr* ➔ pcr electronic excitations, the strength of the metal–metal bond in these complexes should be greater in their dcr*pcr excited states. A 3,5-dimethyl-4-isobutylpyrazole complex was used due to the poor solubility of the corresponding 3,5-dimethylpyrazole complex.
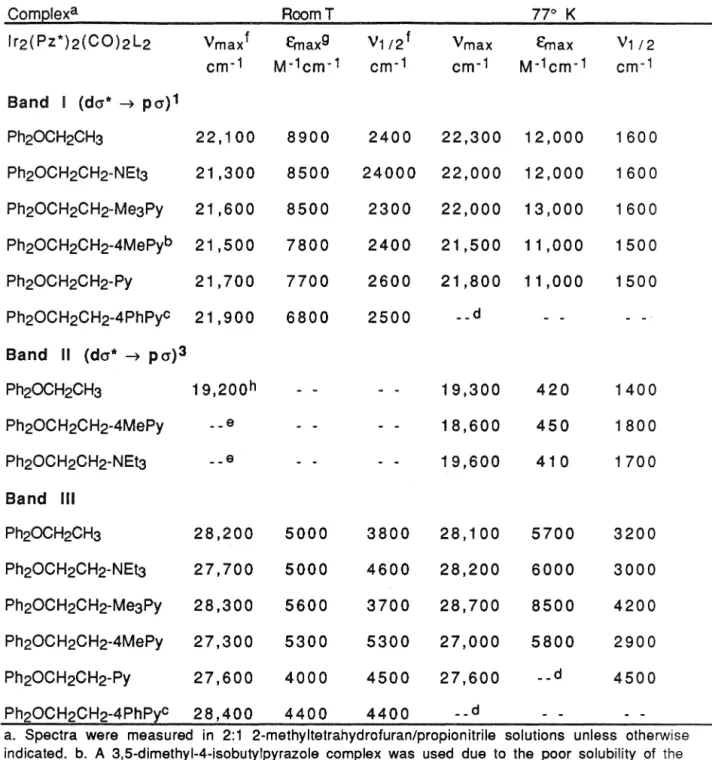
The electronic absorption and emission spectra of the model compounds and donor-acceptor complexes were largely identical within their experiment. Absorption Spectra: Room temperature absorption spectra of donor-acceptor complexes and model compounds in 2:1 2-methylTHF/propionitrile solutions show two intense absorption features with maxima at approximately 21,000 cm-1 (E = 8500) and 27,900 cm - (e = 5000). In terms of the relative intensities and energies of the I, 11 and 111 bands, the electronic absorption spectra of carbon monoxide, dimers of phosphine metals, are remarkably similar to the spectra of a series [lr2(Pz)2(COD)2 ] complexes.
Similar bands appear in spectra of [lr2(Pz)2(CO)4] and its analogues, but are absent in spectra of the corresponding pyrazolate-bridged COD systems.8 These bands have been tentatively assigned to (d1t ➔ p,1t* CO ) transitions based on their 500 cm·1 vibrational progressions and their similarity to features observed in monomer spectra. 12 Their low intensity in spectra of the lr2(Pz*)2(CO)2(Ph2POR)2 systems and absence in spectra of the COD dimers indicate that these are forbidden transitions, gaining significant intensity via. K the singlet and triplet emission intensities of donor-acceptor compounds were similar to the intensities of the lr2(Pz*)2(CO)2(Ph2POCH2CH3)2 and.
Furthermore, the 77°K phosphorescence lifetimes of the donor–acceptor and model compounds were the same within their experimental uncertainties. A 4-isobutyl-3,5-dimethylpyrazole complex was used due to the poor solubility of the corresponding 3,5-dimethylpyrazole complex.
MAVELENGTH
As already mentioned, rigidochromic emission appears only in the spectra of [lr2(Pz)2(CO)4] and. Very interesting is the intensity damping observed in the singlet emission bands of donor-acceptor compounds. A more detailed presentation of electron transfer reactions observed in donor-acceptor systems will be presented in Chapter 4.
The electrochemical properties of the donor-acceptor and model compounds were interesting from several points of view. Identifying this species in time-resolved absorption spectra of the donor-acceptor complexes would be greatly facilitated if it were. Cyclic voltammograms of the electrolysis products differ significantly from CVs of lr2(Pz*)2(CO)2(Ph2POCH2CH3}2.
Donor-acceptor complexes: The cyclic voltammograms of the donor-acceptor compounds were, in general, qualitatively similar. Part a shows all the redox couples seen in the CVs of the donor–acceptor complexes between ±1.5 volts. In contrast to lr2(Pz*)2(CO)2(Ph2POCH2CH3)2, the CVs of the donor-acceptor compounds show three electrochemical pairs in the potential range between + 1.5 and -1.5 volts vs. SSCE.
This couple is assigned to the one-electron reduction of the two covalently bonded pyridinium cations in the donor-acceptor systems. A careful examination of the redox potentials for the donor-acceptor complexes and model compounds reveals an important trend in their electrochemical properties. Characterizing the relative energies of the metal localized (1B and 3B) and charge transfer states (CT) in the donor-acceptor systems is. important to understand their photophysical properties.
In the previous section, it was shown that pyridinium cations change the oxidation potentials of donor–acceptor complexes by acting as inert electrostatic charges.