Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA. Department of Pathology and Laboratory Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, Stellar-Chance Laboratories, PA, USA. Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY, USA.
Department of Pathology and Laboratory Medicine, School of Medicine, University of North Carolina, Chapel Hill, NC, USA. Department of Pathology, ARUP Institute of Clinical and Experimental Pathology, University of Utah, Salt Lake City, UT, USA. Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA.
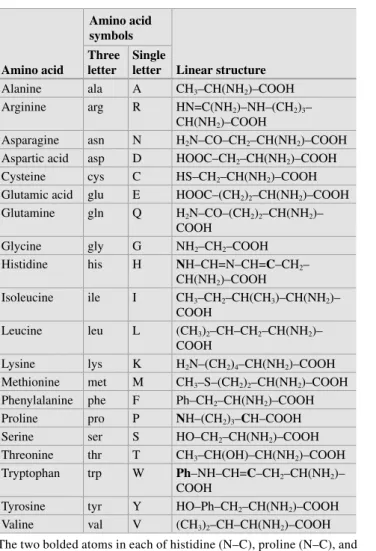
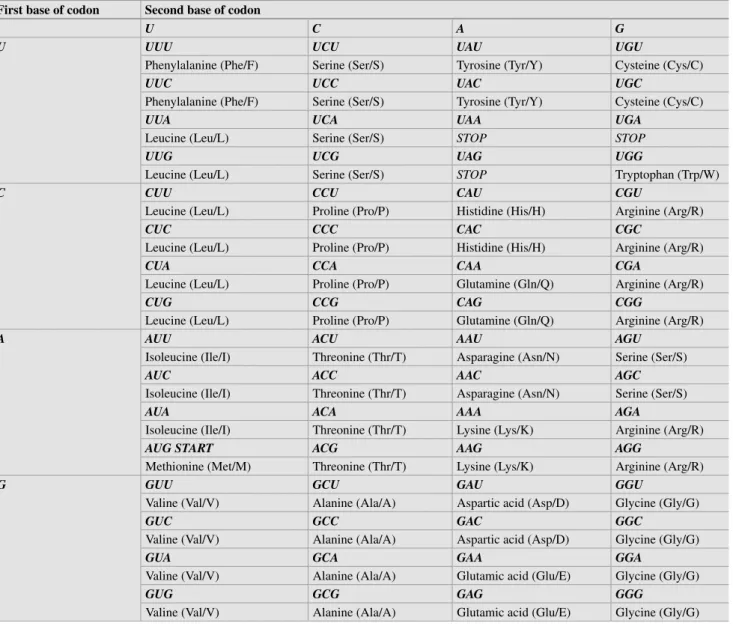
Basics of Molecular Biology
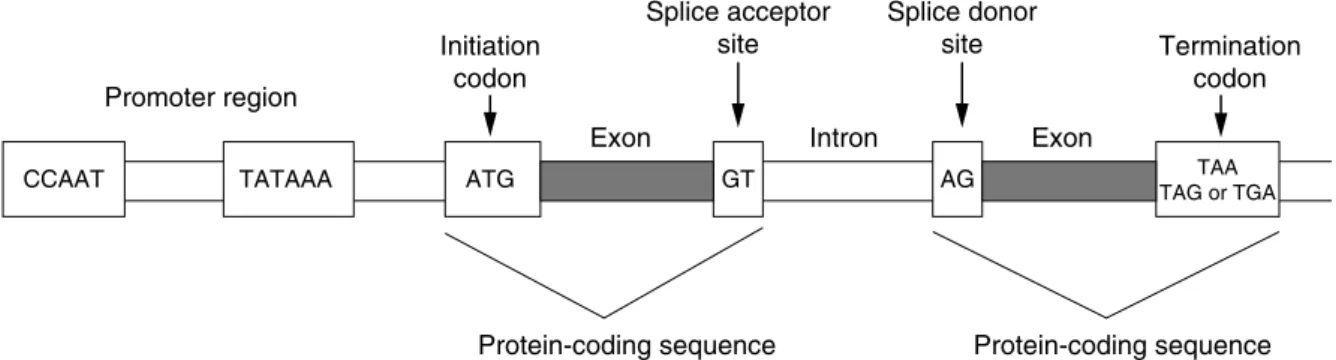
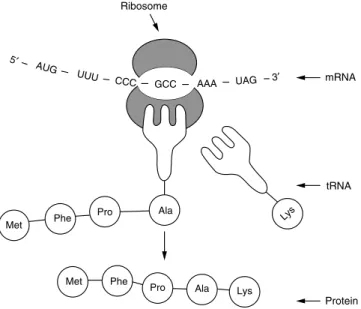
THE TRIPHOSPHATE BOND AT THE END OF THE TRANSCRIPT IS ALSO KNOWN AS A CAP. Mutations in the polyadenylation signal have been associated with altered transcriptional stability in the PROTHROMBIN CASE. FROM THE ANTICODON SEQUENCE WITH THE GROWING POLYPEPTIDE CHAIN AND OCCURRING IN THE SMALL SUBUNIT OF THE RIBOSOME.
PCR is more sensitive than Southern blot hybridization due to the amplification of the target sequence. The genotype is based on the amplification in one of the reactions alone (homozygous non-variant or variant) or both reactions (heterozygous). AS-PCR can detect one variant allele in the presence of 40 copies of the non-variant allele.
The fluorophore is quenched by the complementary structure of nucleotides in the stem of the hairpin. Mutations of the BRCA1 gene in hereditary breast and ovarian cancer in the Czech Republic.
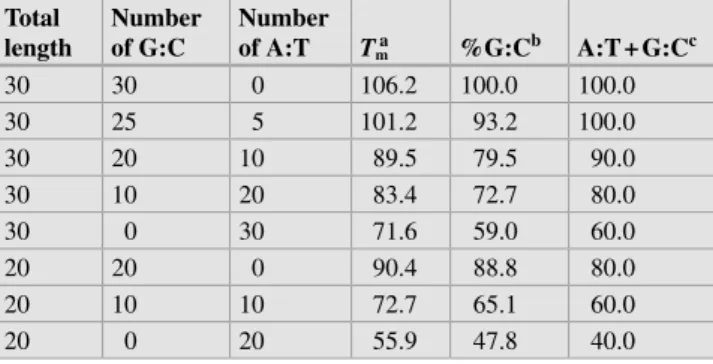
![Table 1.1 #OMPARISON OF SIZES IN BASE PAIRS OF VARIOUS GENETIC elements [n]](https://thumb-ap.123doks.com/thumbv2/123dok/10279968.0/22.892.457.816.119.466/table-omparison-sizes-base-pairs-various-genetic-elements.webp)
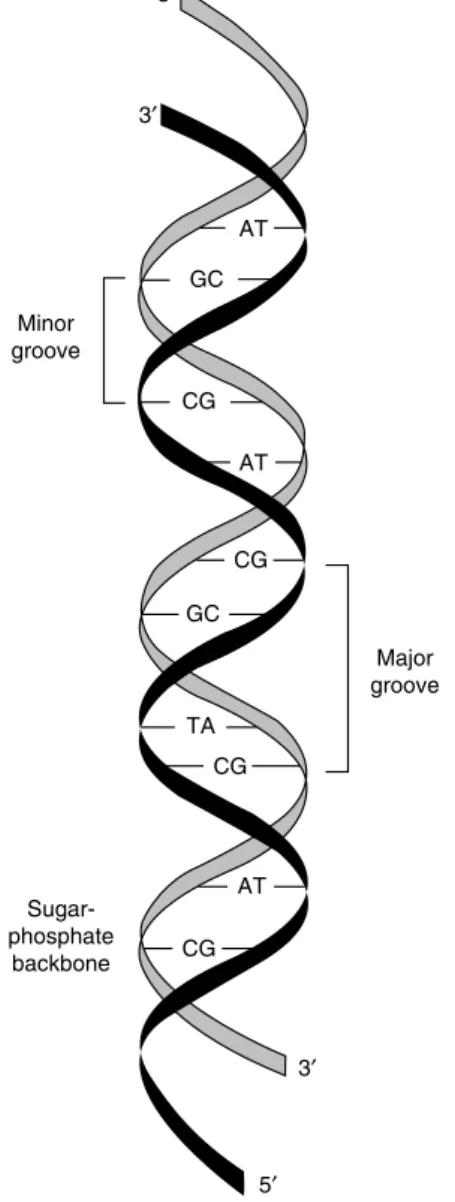
Genetics
Genetic Counseling
Linked Dominant Inheritance
The risk to the offspring of an affected female is 50%, regardless of the sex of the offspring. The risk to the offspring of affected males is sex dependent, with all daughters, but not sons, inheriting the gene mutation. However, many of these conditions are fatal in males, so pedigrees may show an over-representation of females or an increased rate of miscarriage, presumably from affected male fetuses (see Table 4.2 and Figure 4.6).
Linked Recessive Inheritance
Linked Inheritance
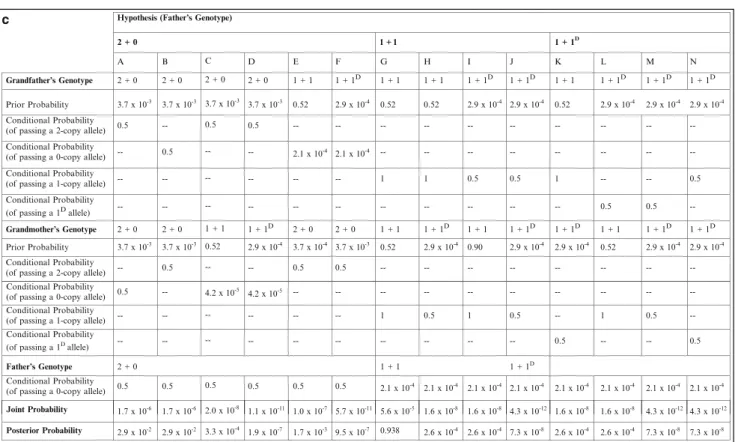
The area of the small rectangle containing three unshaded squares (for three unaffected sons) is 1/8 of the left half and represents the conditional probability of three normal sons under the hypothesis that the consultant is a carrier. The area of this larger rectangle is 1/2 of the total area and therefore also represents the joint probability that the consultant is a non-carrier (1/2) and that as a non-carrier she would have three normal sons (~1 ), or. Likewise, the posterior probability that the consultant is a noncarrier is the area of the larger rectangle with three unshaded squares (for three unaffected sons) divided by the area of the entire inverted-L-shaped box, or.
As in the first case, the joint probability for each hypothesis is the product of the prior and conditional probabilities for that hypothesis. The area of the lower left small rectangle occupies 1/10 of the 2/3 carrier area and represents the conditional probability of a normal test result under the hypothesis that the counselor is a carrier. The area of the rectangle encompassing the entire 1/3rd non-carrier region represents the conditional probability of a negative test result under the hypothesis that the consultand is a non-carrier.
The area of this rectangle is 1/3 of the total area and therefore also represents the joint probability that the consultant is a non-carrier (1/3), and that she will test negative as a non-carrier (~1 ), or. The mt/mt genotype (in brackets) is excluded based on the fact that the consultant is unaffected. c) Bayesian analysis for the consultant in a. d ) Schematic representation of the Bayesian analysis of c (see text). Similarly, the posterior probability that the consultant is a non-carrier is the area of the larger rectangle on the right divided by the area of the inverted L-shaped box, or.
As in the generalized Bayesian analysis shown in Table 5.1, the joint probability for each hypothesis is the product of the prior and conditional probabilities for that hypothesis, and the posterior probability for each hypothesis is the joint probability for that hypothesis divided by the sum . of all joint probabilities. As in the generalized Bayesian analysis shown in Table 5.1, the joint probability for each hypothesis is the product of the prior and conditional probabilities for that hypothesis, and the posterior probability for each hypothesis is the joint probability for . The prior father probabilities are the products of the prior and conditional probabilities for the grandparents for each column/permutation.
Again, the prior probabilities of the father are the products of the prior and conditional probabilities of the grandparents for each column/permutation.
Intellectual Disability
Banded Karyotyping
The limit of resolution of G-banding is between 5 and 10 Mb and depends on the region of the genome and. However, FISH can only detect deletions, duplications and rearrangements of the chromosomal region complementary to the probe. The copy number of some regions of the genome cannot be accurately evaluated by this technology, especially repetitive regions and small deletions and duplications.
Also, arrays will not detect CNVs in the regions of the genome that are not on the array platform, and therefore the resolution of the array is dependent on both the design and the genomic architecture. This is a growing field and reappraisal of the clinical. uncertain findings at a later date may lead to the reclassification of some of these results. A primary consideration for the interpretation of results is the functions and density of the genes within the variant interval.
When assessing the gene content, the impact of gene dosage on the function of the genes in the interval should be taken into account. For deletions, the primary consideration will be on the evidence for the phenotypic effect of haploinsufficiency in the region. For duplications, the primary consideration will be the triplosensitivity of the region; however, duplication breakpoints in a gene may or may not interfere with gene function.
CNV in a clinically normal parent may not exclude a causal relationship between the change and the patient's phenotype. As more medical specialists use this technology, the understanding of the pathogenesis of ID/DD will improve. This deletion is within the SOX5 gene and includes two untranslated exons in the 5' untranslated region of one of the SOX5 transcripts.
Subtelomeric FISH analysis of 11 688 cases: an assessment of the frequency and pattern of subtelomeric rearrangements in individuals with developmental disabilities.
Fragile X Syndrome
Two types of protein glycosylation occur, N-linked and O-linked, which differ in the attachment of the oligosaccharide to protein. Even in the most common CDG subtype, PMM2-CDG (CDG-Ia), individuals with the same mutation have variable severity of the clinical symptoms [12. Some of the undetected mutations may reside in the large dystrophin introns or in regulatory regions. .
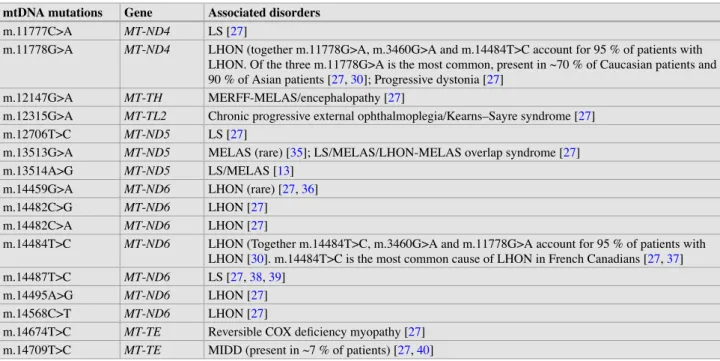
The high rate of de novo mutations in SMN1 may account for the high carrier frequency in the general population, despite the genetic lethality of type I disease. The disease is caused by a CAG trinucleotide repeat expansion in the coding region of the gene androgen receptor (AR) [45. An explanation for the phenotypic differences between patients carrying partial deletions of the DMD locus.
The two ribosomal RNAs (rRNAs) (12S and 16S) and 22 transfer RNAs (tRNAs) encoded by mtDNA are also involved in the translation processes of the mitochondrial genome. Most of the nuclear gene-encoded proteins are synthesized in the cytoplasm and imported into mitochondria. Among the mtDNA mutations in the protein-coding genes associated with other phenotypes are the m.8993T>G and.
Mutations in any of the genes involved in COQ10 biosynthesis can cause CoQ10 deficiency. Although not involved in the structure and assembly of the OXPHOS system, both SLC25A4 (ANT1) and SLC25A3 (PHC) are important for the main function of the OXPHOS system—ATP synthesis. For the majority of patients with a mitochondrial disorder, the causative mutation may be in the mtDNA or in one of the many nuclear genes important for normal mitochondrial function.
The other 2% are caused by defects in the biosynthesis or regeneration of the cofactor of PAH, 6(R)-l-erythro-tetrahydrobiopterin (BH4). MSUD is caused by a deficiency of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex. The overall incidence of the disease is estimated at 1 in 50,000 in the United States, with similar statistics reported in Japan.
Fibroblast Growth Factor Receptor and Related Skeletal Disorders
Ligand binding induces receptor homodimerization and autophosphorylation of tyrosine residues located in the cytoplasmic domain to propagate the intracellular signal. FGFR-associated craniosynostosis and/or chondrodysplasia-causing mutations tend to cluster in specific receptor domains (Fig. 12.2a. Analogous mutations found in each of the different FGFRs tend to reflect phenotypic effects between receptors.
Mutations associated with isolated non-syndromic forms of craniosynostosis have been found in each of the FGFR1-3 genes. Often, full sequencing of the FGFR genes may be requested to rule out the receptors and potentially detect de novo mutations. An activating mutation, p.Pro252Arg in the FGFR1 gene, accounts for approx. 5% of diagnosed patients.
Heterozygous mutations in the FGFR2 gene show high penetrance and variability of the phenotype within families. About half of the cases are inherited and the other half arise from de novo mutations. The mutations associated with ABS include p.Trp290Cys and p.Ser351Cys, both found in the IgIII domain of the FGFR2 gene [72.
Mutations in FGFR1 are found in the conserved amino acids clustered in the C-terminal region of the IgIII immunoglobulin domain, the linker region, and the transmembrane domain. Mutations in the FGFR1 gene can also result in cleft lip and/or palate, agenesis of the teeth and digital malformations [102. A glycine 375-to-cysteine substitution in the transmembrane domain of the fibroblast growth factor receptor - 3 in a neonate with achondroplasia.
Analysis of alternative splicing of the FGFR2 transcript due to a novel 5ƍ splice site mutation (1084+1G>A): case report.