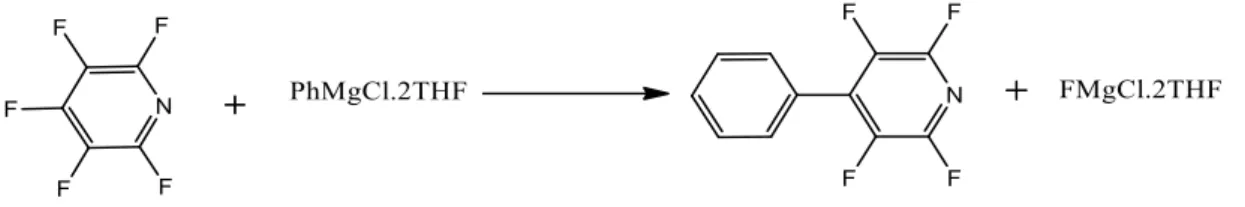
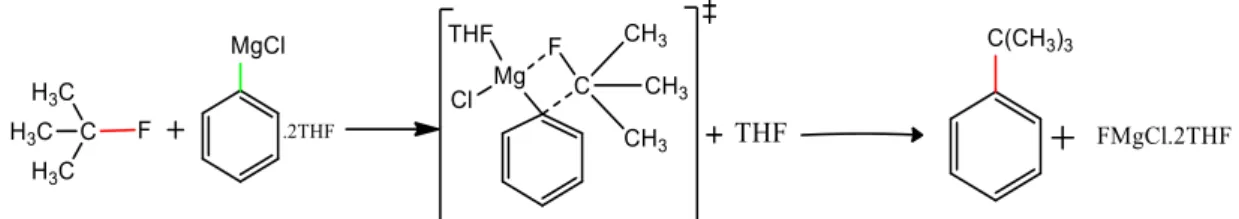
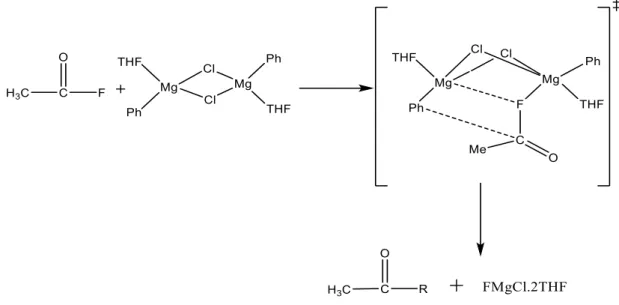
3 Scheme 2: Plausible predicted mechanism for the sp3 C-F bond reaction with PhMgCl, with Mg being monosolvated in the transition state. 31 Scheme 11: Plausible mechanism for C-F activation in the presence of a directing group with a dimerized Grignard reagent.
Introduction
Importance of Carbon-Fluorine bond
The molecular orbital electronic structure of C-F bond has an empty low-lying LUMO σ*C-F, through which it interacts with an unshared pair of electrons. The orbital of lone pair of electrons (lone pair) overlaps with the antibonding orbital of C-F bond and has the antiperiplanar conformation.
Grignard reagents and Schlenk Equilibrium
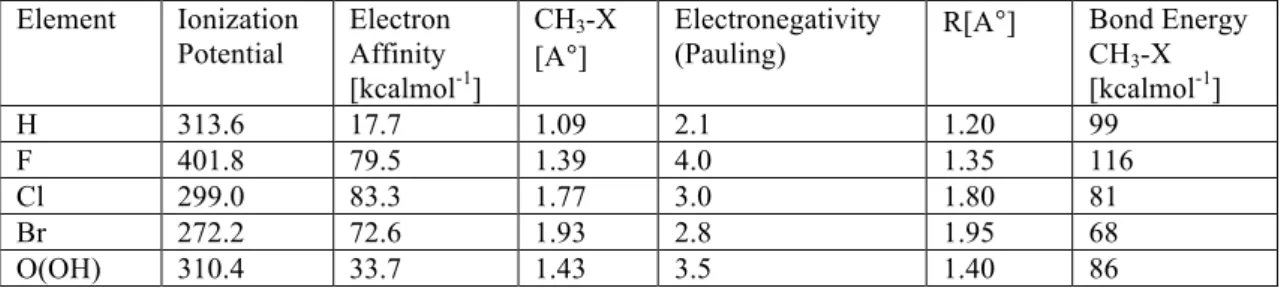
The data in Table 1 clearly show that as the Pauling electronegativity of the halogen atom increases, the bond dissociation energy decreases, which is also consistent with the increased polarity and thus the bond strength. This difference in coordination abilities is due to the Lewis basicity of the solvent molecule.
Organofluorine compounds and its Importance
Various dimeric structures of Grignard reagents are also present in solution; two of which are shown in Figure 1. After this we proceed to the importance of Grignard reagents in synthetic methodology and the reaction of Grignard reagents with compounds containing C-F bond for the formation of C-C bond without transition metal catalyst.
Hatree-Fock Method
The electronic wave function for monoatomic molecules is relatively simple and can be easily calculated, but the problem comes with the electronic wave function of polyatomic molecules, which depends on the bond distance, bond angles, and dihedral angles. The equilibrium bond distances, bond angles, and dihedral angles are then found as the values that yield the minimum electronic energy, including nuclear repulsion. Semi-empirical methods use a simpler Hamiltonian than the correct molecular Hamiltonian and use parameters whose values can be fitted to experimental data or the results of ab initio calculations.
In contrast, the Ab initio (Latin for "from the beginning") method, or first-principles calculations, use the correct Hamiltonian and do not use experimental data other than the values of physical constants.
Density Functional Theory (DFT)
Methods and Basis sets
6-311+G (2d, p) basis set is single zeta for the core atomic orbitals, triple zeta for valence atomic orbitals. We used a lower level HF/3-21G* method for the optimization and B3LYP/6-311+G (2d, p) for the single point energy calculations of the optimized structures to reduce the cost of calculations. We tried two ways of optimizing the structures: (a) optimization with B3LYP/6-311+G (2d, p) and (b) optimization with HF/3-21G* and then single point energy calculation with B3LYP/6 -311 +G (2d, p)29,30 and it was observed that both the results are very close within the error of approximations adopted for the methods.
Furthermore, QST232 (without initial guess of transition state) calculations were used for the prediction of the most likely transition state from the most likely structures of reactants and products.
Transition state
QST2 requires two molecule specifications, for the reactant and product as input, while QST3 requires three molecule specifications, the reactant, the product, and an initial structure for the transition state in that order. The main purpose of our study is to predict the most likely mechanism and to find out the transition state for the reaction of organofluorine compounds with Grignard reagents to form the C-C bond. To carry out this research we have used computational methods to understand the mechanism, HF (HatreeFock) and DFT (Density Functional Methods) have been used to optimize the structures and QST2 (Quasi Synchronous Transit) to predict the transition state. .
A computational study was performed on the plausible predicted mechanism for the reaction between the Grignard reagent and the unactivated sp3alkyl-fluorinated compounds to highlight the mechanistic pathway and to predict the transition state.
Choice of alkyl group
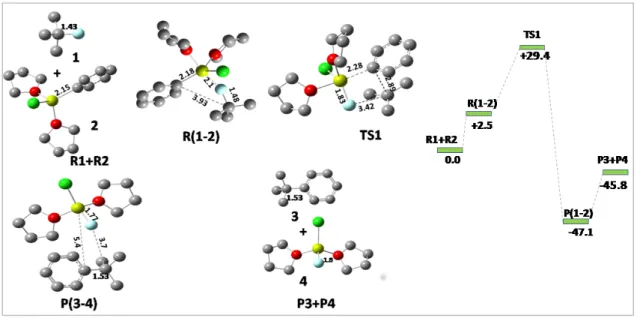
3 Activation of aliphatic C-F bond with Grignard reagent In this chapter we will discuss the activation of C-F bond of saturated carbons ie. we will also emphasize how the reaction proceeds and what all bond parameters change as they undergo the transformation from reactants to products. The analysis is performed on the gas phase reaction and the reactivity of PhMgCl versus CH3MgC. It was observed that alkyl C-F bonds can react with Grignard reagent without the need for activating group and this reactivity is attributed to the relatively longer bond length of sp3C-F bond (1.43Å) compared to sp2C-F bond (1, 35Å), and therefore activation of unactivated alkyl C–F bond with weak Lewis acid magnesium reagents has been successfully achieved in the absence of the transition metal catalyst 34 .
Pathway 1: Mono-solvated Mg
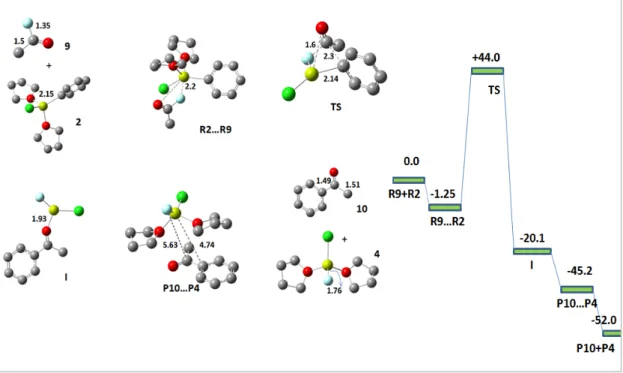
This encouraged us to predict the actual transition state as we go from reactants to intermediates. So, we finally arrived at a transition state for the reaction which is at + 32.6 kcal/mol with C-F, Ph-Mg, C-Ph and Mg-F bond distances of and 1.80 Å, respectively above the reactants. The above reaction follows a stepwise mechanism with overall reaction being exothermic and the reaction proceeds via a transition state for which an activation barrier must be overcome. In the reaction pathway, the alkyl fluoride first undergoes rehybridization and then pyramidal inversion to form the substituted product.
In the reactant, the alkyl fluoride is sp3-hybridized and the magnesium in the Grignard reagent is tetrahedrally coordinated, but as it goes from the reactant to the transition state, the sp3 carbon becomes planar and is now sp2-hybridized with the C-F bond distance changing from 1.43 to 2.89Å.
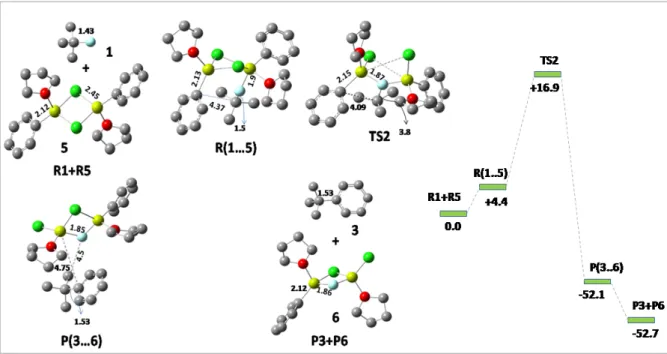
Pathway 2: Disolvated Mg
As can be seen that when Mg 4 is coordinated (Scheme 2) C-Ph bond distance is 4.16Å which is still high for an interaction to be made, while when Mg 5 is coordinated (Scheme 3) this C-Ph bond distance takes relatively down and is now 2.89Å which is more realistic for a bond to form. Therefore, it can be concluded that second mechanism is the actual mechanism while undergoing the reaction. The reaction goes directly to the product from the transition state and no intermediate is involved.
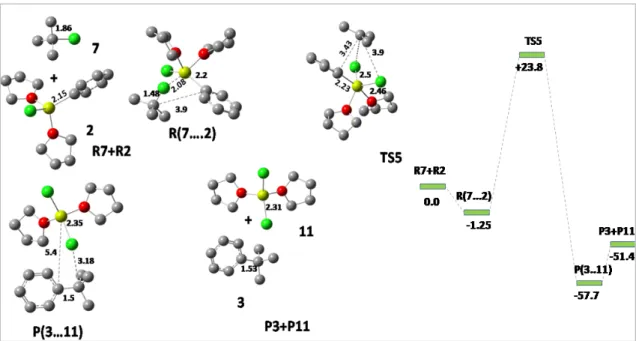
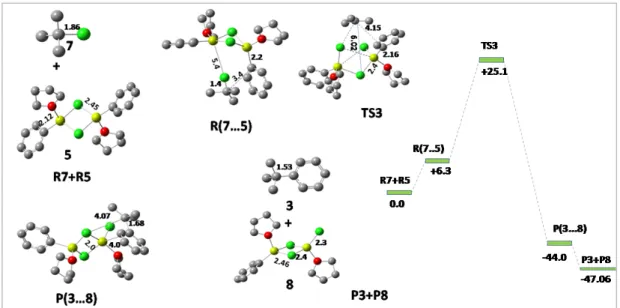
Pathway3: Dimerization of Grignard reagent
Now, the alkyl C-F bond approaches the Grignard reagent and where two main interactions occur; one is between Mg-F and the other is between C-Ph and a transition state begins to form. In the transition state, fluorine acts as a bridging atom between two Mg atoms with the Mg-F bond distance of 1.87Å, each Mg atom remaining tetrahedrally coordinated. The reaction was observed to proceed via the linear dimer pathway rather than the cyclic dimer pathway traditionally followed via a 4-membered cyclic transition state, with the reactive alkyl group being planar.
Reaction of sp 3 C-Cl with PhMgCl
Monomer mechanism
Here again, we investigated the transition state for the reaction, but with a monomeric mechanism where Mg is bound to two solvent molecules in the transition state. To carry out this exchange, the length of the Mg-Ph bond increases, and at the same time the C-Ph bond begins to form and the carbon at the reaction site becomes flat to facilitate attack. Since the C-Cl bond is easy to break compared to the C-F bond due to the relatively longer bond length in the former, it was also found that the reaction barrier is relatively less in C-Cl transformation to C-C bond formation as compared to C-F bond transformations.
After this step, the reaction proceeds smoothly with the complete separation of the two products giving the final release of energy.
Dimer Mechanism
This can be predicted due to low bond strength of C-Cl bond, whereby chlorine atom is readily transferred to electropositive magnesium atom. Therefore, it can be concluded that the C-C bond formation from C-Cl bond occurs via the monomer mechanism instead of dimer mechanism.
Reaction of acetyl fluoride with PhMgCl
Monomer mechanism
The reaction mechanism for the gas phase reaction between acetyl fluoride and PhMgCl/CH3MgCl to form ketone as product is studied. First, a reaction pathway was predicted (Scheme 7), and then the optimizations and energy calculations were performed using the same methods and basis sets as mentioned above. The product was stabilized by -56.47 kcal/mol energy with PhMgCl as well as with CH3MgCl, indicating that the reaction is equally likely with both PhMgCl and CH3MgCl.
To confirm the transition state, we performed a QST2 calculation and obtained exactly the same transition state as predicted in the single imaginary frequency mechanism, with a bond length of Mg-O 3.06, Mg-F 1.87, Mg-R 2.14 Å, this clearly shows that when Mg enters an environment with a more electronegative element, it begins to coordinate with fluorine, and at the same time the R-Mg bond weakens to facilitate the formation of the C-C bond.
Dimer mechanism
In this chapter we will look at how a C-F bond can be activated for the coupling reaction with Grignard reagent, creating a C-C coupled product. We will also understand why an sp2 carbon cannot be activated without the help of a driving group, thus which groups can act as a driving group in this type of reaction. The two systems we will study here are the activation of C-F bonds on the pyridine ring, the pyrimidine ring and in which pyridine acts as an ortho-directing group for the activation of the C-F bond.
Discussion
We will also understand why a sp2 carbon cannot be activated without the help of a driver group, so what.
Analysis of results
In the second step, the nucleophilic attack of phenyl ring from Grignard reagent goes to the reactive site, i.e. after this step, the reaction again proceeds via a confined state, and this structure is the second transition state in the reaction mechanism. In addition, the reactive carbon is now sp3-hybridized with four substituents as shown in the figure. A perfect six-membered (open book) ring is formed in the transition state.
The pyridine ring rotates in the opposite direction, which is an important condition for the stabilization of the products.
Dimer mechanism
Dimerization of the Grignard reagent is a consequence of the electron-deficient nature of the magnesium atom. We have already explored the first case and here we have calculated the stabilization energy for the second path.
Activation of C-F bond on pyridine ring
- Para Product
- Ortho product
- Meta Product
- Dimer Mechanism
In the para-substitution of the fluorine atom with phenyl ring, the reaction proceeds via an energy barrier of +18.82 kcalmol-1 above the reactants. The orientation of the pyridine ring in the structure clearly indicates that it enjoys no coordination with the magnesium atom. In this case, the reaction barrier was found to be +43.9 kcalmol-1 above the reactants and the stabilization energy of the reaction was -69.02 kcalmol-1.
The third and final possibility that we have checked here is the reaction at metacarbon of the pyridine ring, i.e. nucleophilic substitution of a fluorine atom by a phenyl ring and the formation of the product.