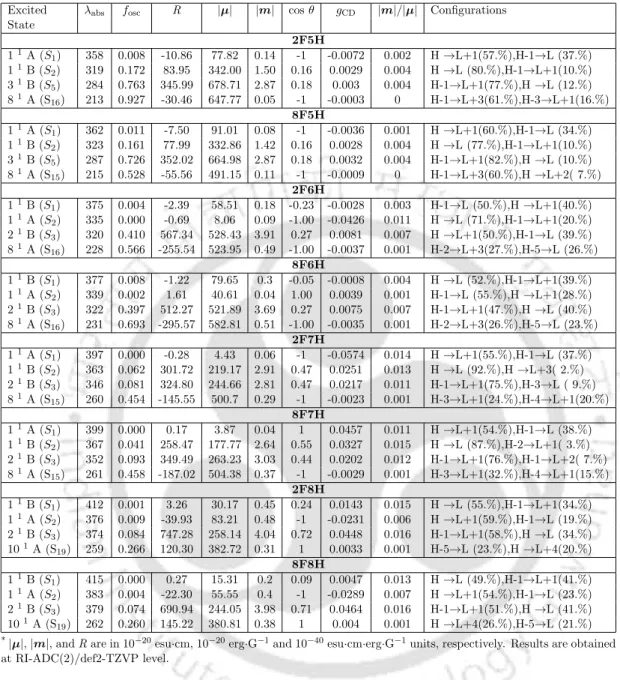
In the next chapter, chiroptical properties of 9Ha and its three derivatives are reported. C9 Excitation Energies (λabs, nm), Corresponding Oscillator Strength(fosc), Orbitals involved in the transitions,|µ|,|m|, cosθ,R,gCDog|m|/|µ|of 8F6H.|µ|,| m|, andR are in esu·cm, erg·G−1 and esu·cm·erg·G−1 units, respectively.
Types of helical-shaped systems
Single-stranded helical systems
- Ortho-substituted ring systems
- Fused-ring systems
In the recent past, many chiral molecules with good absorption and especially luminescence properties have been designed. Small changes in the structures of the helical systems, ring substitutions and the construction of double/triple helicenes have recently been used to achieve large gCPL values.
Multi-stranded helical systems
- Double helicenes
- Triple helicenes
- Quadruple and larger helicenes
In addition, [S]-shaped double aza[6]helicene has also been reported by Tanaka et al.82 Unlike carbo[5,6]helicene, no [S]-type double carbo[7]helicene in anti- form has been reported to date. For example, Itami and co-workers100,101 synthesized five-helical carbo[6]helicenes and five-helical dekathia[6]helicenes.
Motivation
In 2013, Dichtel et al.97 synthesized extended and partially fused quadruple carbo[5]helicene as a π-elongated derivative of double[5]helicene70 (i.e. hexabenzoperylene). It contains four carbo[5]helicene units and none of these four units share a ring among themselves.
Thesis outline
MI is the mass of the nucleus, and ZI and ZJ are the atomic numbers of the nuclei I and J, respectively. Here, the first and second terms correspond to the kinetic energy of electrons and nuclei, respectively.
Born-Oppenheimer approximation
The One-Electron wavefunction and Hartree-Fock method
Post-Hartree-Fock methods
Møller-Plesset perturbation theory
The first-order correction to the energy obtained after the operation of the perturbation operator is given by Therefore, the finite matrix elements in the case of the second-order correction are obtained only for double excitations, as shown in Eq.
Density functional theory
Exchange-correlation functionals
Separate-range hybrid functionals produce more accurate properties of the excited states compared to the most commonly used standard functionals, especially when charge transfer excitations are involved. The parameter α corresponds to the HF exchange contribution over the entire range by a factor α, and β corresponds to the DFT counterpart over the entire range by a factor 1-(α+β).
Dispersion-corrected DFT
The first and second terms take into account the short- and long-range interactions, respectively. The additional flexibility resulting from these two parameters makes it possible to verify the importance of the HF exchange contribution for the short-haul region and the DFT counterpart for the long-haul region.
Time-dependent density functional theory (TD-DFT)
This equation shows the exact representation of the linear response theory, and self-consistent solution of this equation can produce the response function χ of an interacting system.
Algebraic diagrammatic construction scheme for the polarization propagator 21
Accordingly, while ADC(1) indicates the first order of perturbations, which is equivalent to CIS, ADC(2) indicates the second order. The rotational power (R) is the imaginary part of the result of the scalar product between the electric dipole transition moment (EDTM) and the magnetic dipole transition moment (MDTM) between the ground and excited states.
One-electron transition density matrix
- Charge transfer number (Ω AB )
- Total charge transfer (ω CT )
- Exciton size (d exc )
- Participation ratio of NTOs (PR NTO )
This graph is known as the Ω graph or the electron–hole correlation graph.165 In the case of two fragments, this is simply a 2×2 graph (as shown in Figure 3.9 in Chapter 3). Later in the section, we provide a quantitative comparison of the excited state properties by calculating some excited state descriptors.
Computational details
All these TD-DFT calculations used the optimized geometries obtained at the RI-MP2/def2-SVP level. Optimizations of the optically active (PT)3-(PT)5 in their S1 states were performed in the TD-CAM-B3LYP method using the same set of basis sets for investigating the luminescence properties.
Results and discussion
- Structures of oligomers and first vertical excitation energies
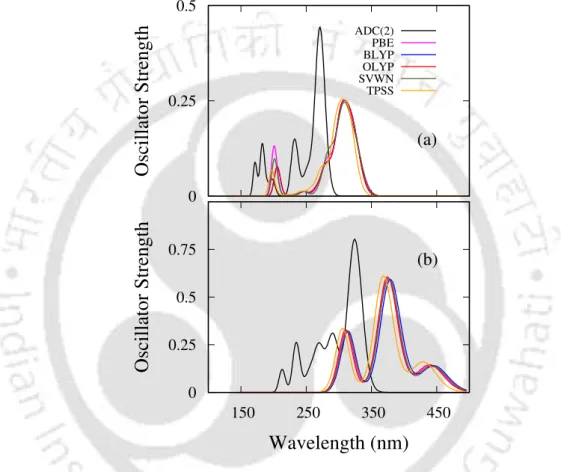
- Absorption spectra
- CD spectra
- Analysis of excited states
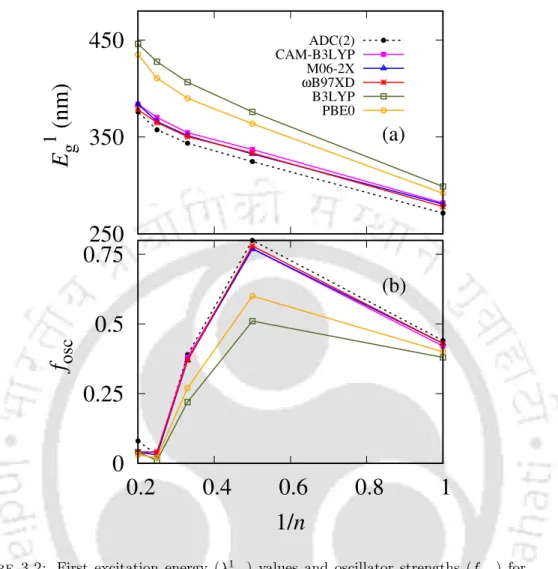
As shown in Table 3.2, the absolute values of R produced by the CAM-B3LYP and lc-PBE functionals are larger than the ADC(2) results for all states except S3 in CAM-B3LYP and. The CAM-B3LYP results shown in Table 3.3 for (PT)5 differ from the ADC(2) results in the PRNTO case. The results of the M06-2X and ωB97XD functionals are very close to the CAM-B3LYP results for all oligomers as observed in the UV and CD spectra.
For (PT)1, differences in the values of the descriptors between these two and a functional like CAM-B3LYP are small. In the case of PRNTO for the S1 state, ADC(2), CAM-B3LYP and lc-BLYP results remain close to each other up to n=4.
Emission properties
Conclusions
For excited-state properties such as UV-vis and CD spectra, the performance of different DFT functionals is evaluated by comparing the results with the ADC(2) results. On the other hand, quinoxaline-based 9HaQ is found to be the worst CPL emitter with the lowest dissymmetry factor. In addition to carbo[n]helicenes, hetero[n]helicenes, which have aromatic rings with heteroatoms in the main skeleton, have also been investigated recently.
As mentioned in the previous section, many Qx-based helicenes have been recently synthesized and chiroptical properties studied. Similarly, many pyrazine-fused helicenes are also reported in the literature.241-244 Moreover, extension of the helix length has also been demonstrated. to improve results in many cases. With the above in mind, our goal is to investigate the effects of introducing these two aromatic acceptor rings and increasing the helix size on the chiroptical properties such as gCD/gCPLandKf. Typically, the magnitudes of m are very small for π-π∗ transitions in most organic molecules.28,66 Therefore, there are a large number of studies in the literature to maximize the |m. With the above in mind, DFT-based calculations are performed to investigate the absorption, CD and CPL properties of these three systems and compare these results with the values for 9Ha.
Computational methodology
In these scenarios, range-separated functionals such as CAM-B3LYP and ωB97XD have been shown to be a better fit. In our case, to verify the applicability of DFT functionals, vertical excitation calculations were also performed by second-order algebraic diagrammatic construction. diagrams of the polarization propagation level ADC(2)152,153 using the same basis set. In TD-DFT and RI-ADC(2) calculations, dichloromethane (DCM) solvent was used using integral equation formalism-polarizable continuum model (IEF-PCM)259 and conductor-like screening model (COSMO)260,261 model, resp. For studies of directions and densities of transition dipole moments, Multiwfn v3.8 software263 was used.
Results and discussion
Ground state structures
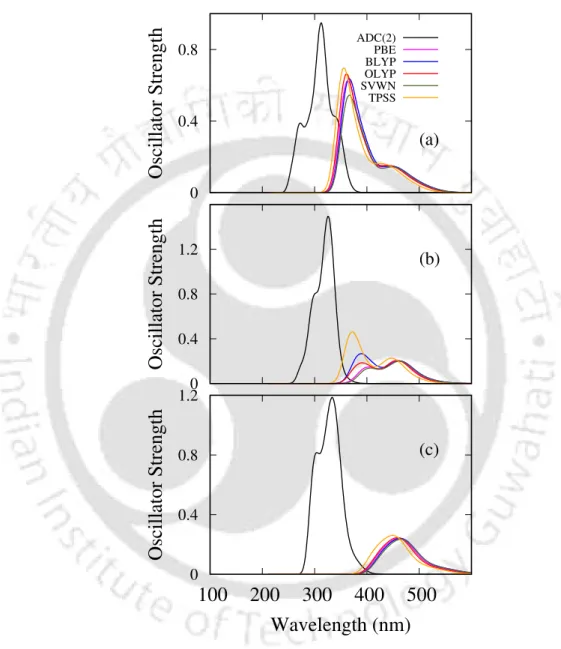
- UV spectra
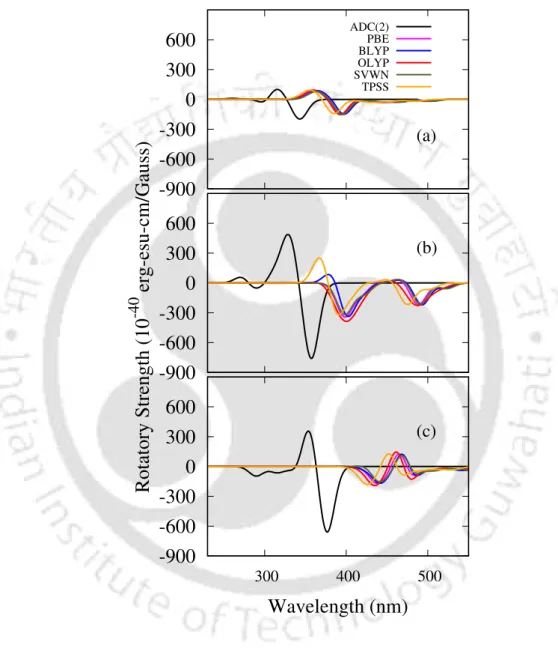
- CD spectra
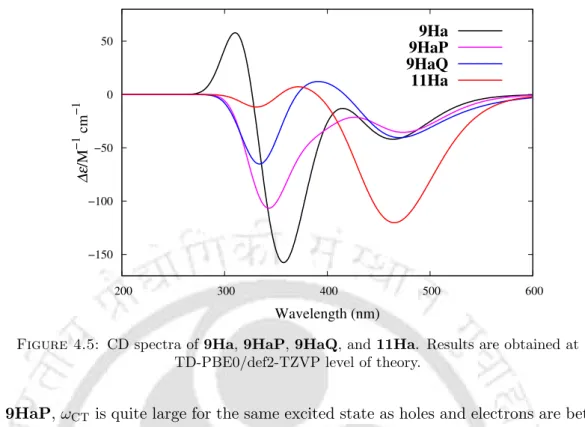
On the other hand, the S0→S6 excitation carries the highest value of fosc and R in the PBE0 and B3LYP results, similar to the values obtained in the ADC(2) case. The peaks at 356 nm (s fosc = 0.248) in PBE0 are in good agreement with the experimental value of 342 nm.11 For the emission in 9Ha, the optimization was performed in the S1 state using the PBE0 functional. In the case of 11Ha, the hole/electron distributions in the S1, S2, and S6 states are similar to those in 9Ha.
Like 9Ha, the transition corresponding to the lowest energy band shows a maximum gCD value of -0.061, while the value decreases to -0.011 in the case of the S0 → S7 transition. In the case of 11Ha, one negative peak with maximum R appears at 462 nm, corresponding to the S0→S1 transition (see Table 4.2).
Excited-state structures and chiroptical properties
As shown in Table 4.1, in all four cases one terminal unit is tilted 20-27° more outward than the other terminal unit, corresponding to the ground state results. However, there is an increase in the value of |µ| in case of emission and this results in a decrease in dissymmetry factor for S1→S0 compared to S0→S1. Qx-fused polyaza[5]-helicene and 7HQ show smaller ΦFL values compared to the respective parent systems.112 However, 9HaQ shows larger Kf compared to that of parent 9Ha as well as 9HaP.
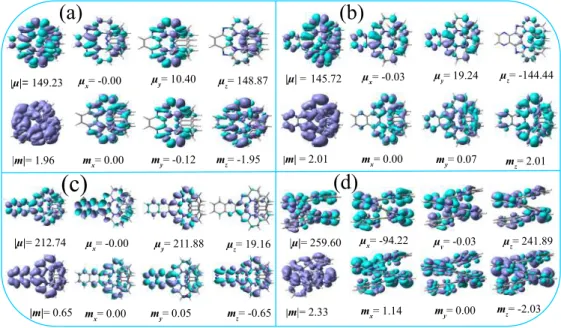
In 11Ha, electron delocalization over a larger area (as shown in Figure B7) leads to the largest values of |µ| and |m|. While the terminal rings contribute primarily to the X- and Z-axis components of EDTM (the densities in the 5-6-5 unit cancel each other), 5-6-5 and its adjacent rings participate for MDTM components.
Conclusions
In typical organic molecules, |µ| much larger than|m|which leads to small g-factors and the values are usually in the range of This value is far away from the maximum value of ±2. As mentioned in the previous chapter, typical modifications are based on either substitution of hydrogens in helicenes with other atoms/groups or fusion of aromatic rings on the helical backbone to produce ring-fused carbon[n]helicenes7,58-63 so far. Similar to pristine carbohelicenes, ΦFL was also found to decrease for substituted carbohelicenes with increased chain length.54,57 Halogenation of carbohelicene rings is another strategy to tune the chiroptical properties.51,55 Synthesis of various fluorinated carbohelicenes has been reported in the literature in the work of Nakai et al.51 observed a systematic enhancement in CD intensities of the 1Bb band together with their corresponding dissymmetry factors after substitutions with F, Cl and Br atoms.
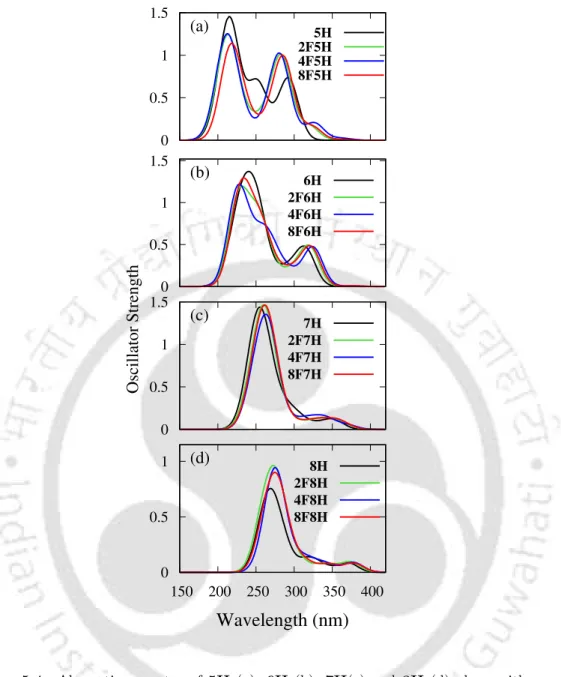
While the specific rotation of a fluorine-substituted compound was found to be less than the value for the parent system, ΦFL was found to increase with the increase in the number of fluorine atoms. The hydrogens at the end rings are replaced by fluorine atoms to produce di- (replacement of the hydrogens with fluorine atoms in the 1- and 10-positions), tetra- (replacement of the hydrogens with fluorine atoms in the 1-, 10-, 4-, and 40-positions) and octafluoro- 6H (replacement of all four hydrogen atoms by fluorine atoms at . each terminal unit).
Computational methodology
The method was found to outperform MP2 for non-covalent interactions such as in cases of π-stacked systems.277–280 The default scaling parameters, cos=6/5 and css=1/3, were used in our studies. Vertical excitation energy calculations at the ground state optimized geometries were performed at ADC(2)152,153 scheme level. Geometry optimizations in the lowest singlet excited states (S1) of all sixteen systems were performed in gas phase at the same level of theory.
The methods used in this study are therefore denoted as RI-SCS-MP2 and RI-ADC(2), for the ground and excited states, respectively. UV and CD spectra were obtained using the Gabedit software.233 Excited state characterizations were performed using the TheoDORE v2.4 software.264,265 Analysis using TheoDORE is based on the 1-TDM.
Results and discussion
- Ground-state structures, absorption and CD spectra
- UV spectra
- CD spectra
- Structures in the S 1 states and emission properties
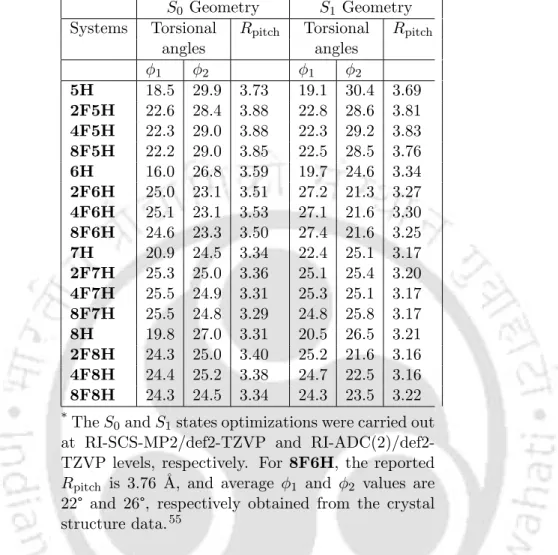
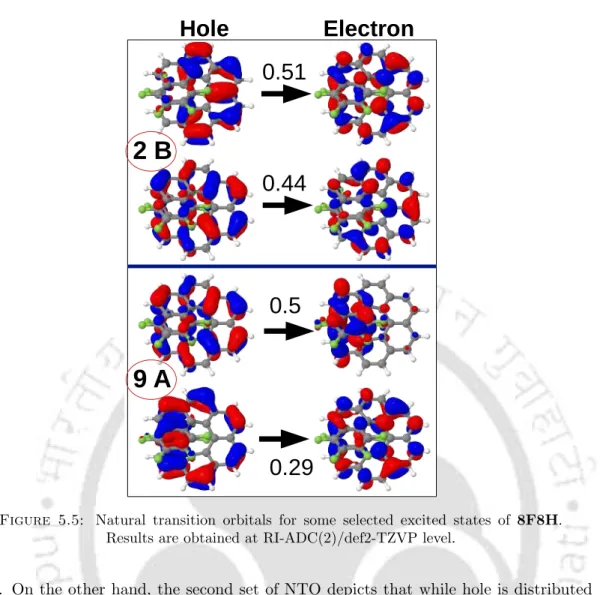
However, the positions of the peaks are slightly red-shifted (by ≈2-5 nm, as shown in table 5.2). The peak in the high energy region (at 261 nm) arises due to the same set of excitations as in the case of 2F7H. In the case of the dominant NTO for 3 1B, CT occurs from the terminal rings and rings adjacent to the terminal rings.
However, a drastic decrease in R value is observed in the case of transition to the nearly degenerate 2 1A state. In the case of 6H, Rpitch for S1 is smaller by 0.25 ˚A compared to the value in the ground state.
Conclusions
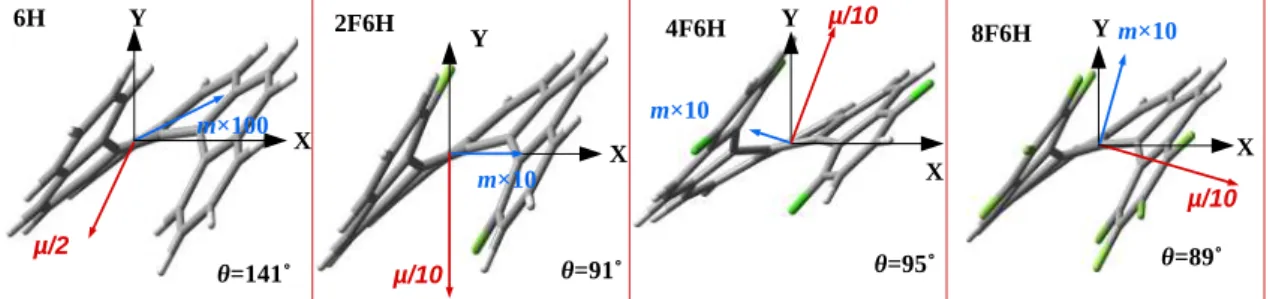
![Figure 5.7: Variations in magnitudes of electric dipole transition (|µ|) and magnetic dipole transition moments (|m|) for carbo[5-8]helicenes and their fluorinated derivatives](https://thumb-ap.123doks.com/thumbv2/azpdfnet/10428016.0/115.893.176.735.112.852/variations-magnitudes-electric-transition-transition-helicenes-fluorinated-derivatives.webp)