I certify that this project report entitled "FIRST PRINCIPLES STUDY ON ALASXSB1-X ALLOYS" prepared by NATALIE TAN WEE SIAN has met the required standard of submission in partial fulfillment of the requirements for the award of Bachelor of Science (Honours) in physics. at Tunku Abdul Rahman University. The copyright of this report belongs to the author under the terms of the Copyright Act 1987 as qualified by the Universiti Tunku Abdul Rahman Intellectual Property Policy. Appropriate acknowledgment should always be given for the use of any material contained in or derived from this report.
The work carried out in this project aims to study the composition dependence of the energy gap values, e.g. of the ternary alloy composites. The ground state electronic properties of the AlAsxSb1-x semiconductor alloy composites were investigated using first-principles calculations of Density Functional Theory (DFT) within the General Gradient Approximation (GGA) functional framework with the implementation of Perdew–Burke–Ernzerhof ( PBE) exchange-correlation (XC). It was shown that there is a non-linear behavior in the composition dependence, x of the band gap values for the ternary alloys, indicating the deviation from Vegard's law, with clear downward bending of the fundamental energy gap values obtained relative to the alloy. fraction composition.
There is a clear indication of the indirect band gaps via the CBM along the L and X paths of the band structures as well as the distinct energy gap dependence on the arsenic composition, x. Recommendations for further investigation include additional crystal visualizations of supercell structures matching that of AlAs0.56Sb0.44, which also have similar BZ structures to that of the constituent binaries, identification of the critical x value where transition from indirect to direct band gap occurs and further attempt to perform calculations that provide results for AlAsxSb1-x random alloy structures.
INTRODUCTION
Introduction to III-V Semiconductor Alloys
A ternary semiconductor alloy consists of a combination of three semiconductor elements, the product of which is a novel composite semiconductor alloy material that has its own unique structural, optical, and electronic properties. Binary alloys can be made with other III-V compounds, giving an intermediate ternary alloy, in this case the focus of this project, AlAsxSb1-x, can be simulated by combining aluminum arsenide (AlAs) and aluminum antimonide (AlSb). binary files. The use of III-V ternary alloys is essential in many optical and electronic devices as they can be optimized and pre-configured to meet specific application requirements, i.e. the potential tunability of the energy band gap depends on the composition and structural configuration of the alloy (Adachi, 2017).
Problem Statement
In retrospect, semiconductor material junctions have long been the subject of research and study due to the ability to adjust the size of the forbidden energy gap within its band structure. Along with the ability to influence other material parameters, these connections can be optimized for application in a wide range of semiconductor devices. The results obtained by varying the alloy compositions, geometry, doping concentrations, lattice strain and feature size maximize the stability of the integration of systems of small superlattices and quantum well structures in device applications (El Haj Hassan et al., 2010).
Furthermore, very limited experimental work has been performed on AlAsxSb1-x ternary alloys due to complications regarding computational resources and challenging calculations related to configurations, arrangements and general disorder within the alloy structures. The combination of AlAs and AlSb constituent alloys to form AlAsxSb1-x alloy composites produces intermediate properties unique to the ternary alloy. Thus, the focus of the study in this project is to study and analyze the compositional dependence of the energy band gap values.
Furthermore, it was found that varying the composition of its constituent components within the crystal structure of the alloy results in a tunable value of the band gap, which could enable the fabrication of materials tailored to the required electronic specifications. The study and analysis of the AlAsxSb1-x ternary alloy not only contributes to the lack of theoretical studies on the alloy material, but could also facilitate further research and future work on material modeling of other quaternary alloys such as AlxGa1-xAsySb1-y, which presents opportunities in.
Aim and Objectives
Scope and Limitation of the Study
Electronic band structures were not mapped for some ternary alloy configurations due to significant distortions in the BZ. Therefore, the accuracy of the energy gap values derived from density of state is uncertain. In addition, it is also unclear whether the geometry of the alloy crystal structure, cell matrices, the size of the alloy supercells, general orientation of the configurations, and apparent BZ distortion have any appreciable effect on the gap values obtained in this study.
Random alloys were not included as a more comprehensive analysis with interdigitated alloys due to the extreme computing resources and time required to complete. In order to simulate sufficiently isotropic random configurations of alloys, the supercell sizes must be large enough. Attempts to perform calculations on a group of random configurations of 3 × 3 × 3 supercell alloys not only yielded results that conflicted with experimental values, but were also not completed in time.
Importance and Contribution of the Study
Outline of the Report
INTRODUCTION
CONCLUSION a. Conclusions
- Introduction
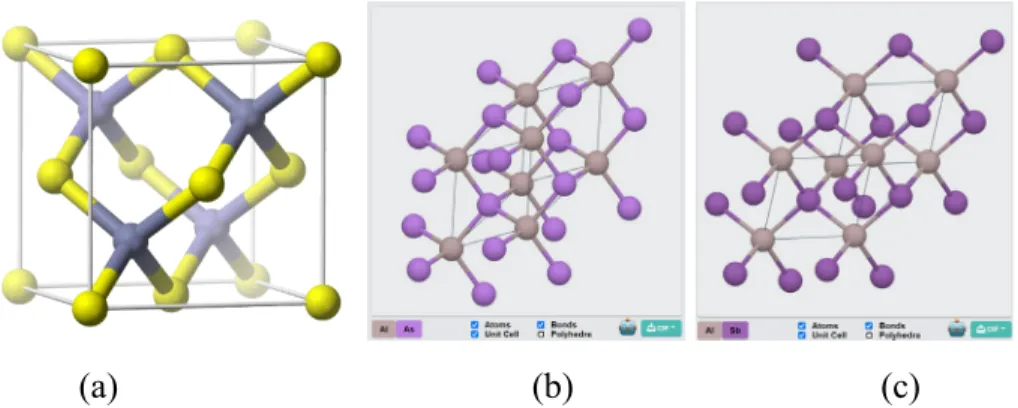
- Aluminium Arsenide, AlAs
- Aluminium Antimonide, AlSb
- Aluminium Arsenide Antimonide, AlAs x Sb 1-x
- Density Functional Theory
- The Interpolation Scheme
- Vegard’s Law
- Bowing Parameter, b
- Introduction
- Materials Modelling with Quantum ESPRESSO
- Crystal Structure Visualization with VESTA and XCrySDen VESTA, which is an acronym for Visualization for Electronic and STructural
- Performing Electronic-Structure Calculations
- XC Functional and Parameters
- Work Plan
- Work Stages
- Introduction
- Electronic Properties of AlAs x Sb 1-x
- Conclusions
- Recommendations for Future Work
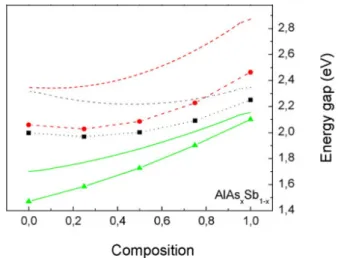
In addition, there are clear variations of nonlinear increase of the indirect energy gap values, particularly along the Γ, X and L k paths relative to the arsenic composition, x in the zinc blende structure of the ternary alloy configurations. To account for the discrepancies for the ternary alloy composites, experimental values of the energy gap of the constituent binaries have been fitted to the calculated arc parameters of the energy gap obtained (see Figure 2.4). By implementing the linear interpolation scheme, equation 2.12 allows the calculation of the ternary parameter, T which is based on the binary parameters for the structure of the alloy in the form of 𝐴𝐴𝑥𝑥𝐵𝐵1−𝑥𝑥𝑆𝑆, since 𝑇𝑇 ≡ 𝐵𝐵𝐵𝐵 𝑋𝑋 and 𝑏𝑏 ≡ 𝐵𝐵𝐴𝐴𝑋𝑋− 𝐵𝐵𝐵𝐵𝑋𝑋.
Thus, an additional term is included as a function of the composition of the alloy composites to address the non-linearity of the energy gap values. Assuming the ternary alloy composite of ABxC1-x, with varying shared composition, x, if x is given as 0.5, the overall band gap diffraction coefficient is thus indicative of the change in the energy gap values, given in the following Equation 2.18. The following expression, equation 2.25, includes the arc parameter in the calculation of the bandgap energy of alloy composites with variable As concentration, x.
In combination with the available external computational resources, visualizations of the binary and ternary alloy composites were performed using a personal computer. The same process was applied for the ternary alloy configurations, with the exception of the k-point visualizations and band structure calculations. The supercells of the AlAsxSb1-x alloy compositions were constructed in all possible interlocking non-identical configurations.
XCrySDen software version 1.5.60 was used to obtain a convenient 3D visualization of the alloy composites, as shown in Figure 3.9. This was done to map the k-points of the band structure during preliminary investigation of the binary alloys. The first phase involves the preliminary calculations of electronic properties of the binary alloys AlAs and AlSb.
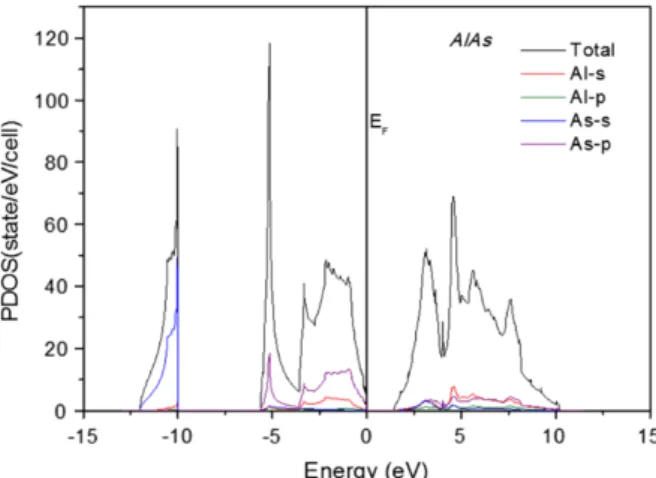
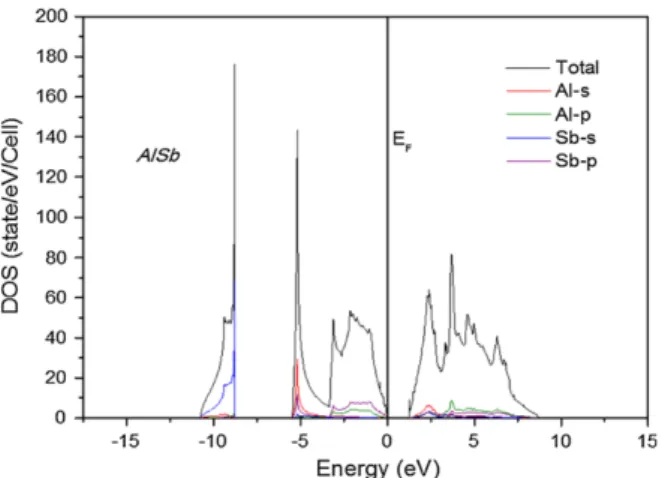
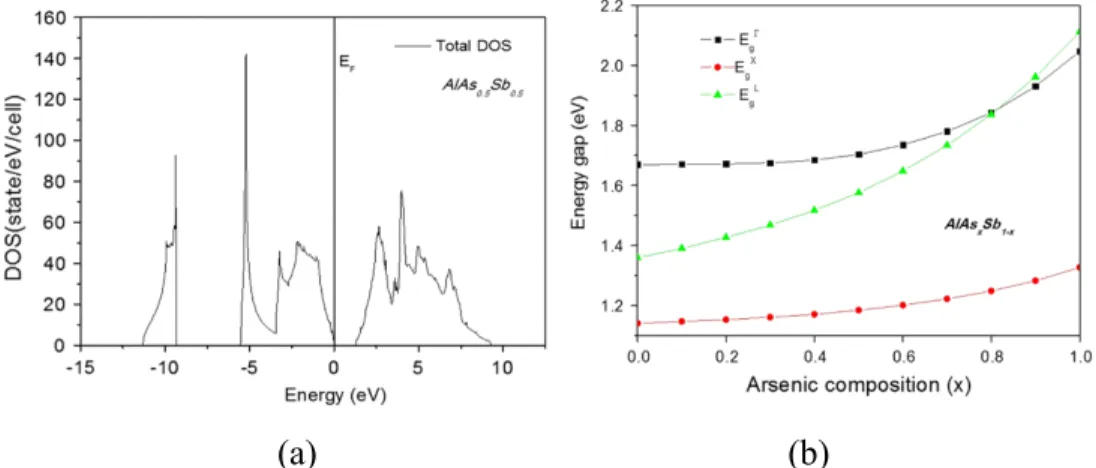
It can be observed from DOS of the ternary alloys that the valence band consists of three separate sections (Bounab et al., 2017). The energy band gap values are derived from the DOS and calculated Fermi energy level of the alloy configurations. Energy band gap values calculated for all the alloy compositions were fitted with a polynomial curve and analyzed against Vegard's law,.
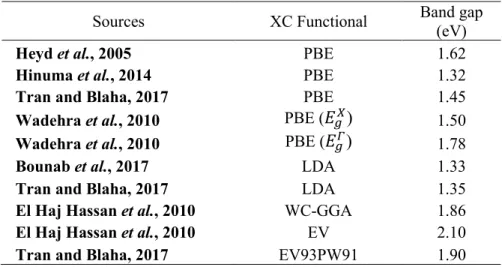
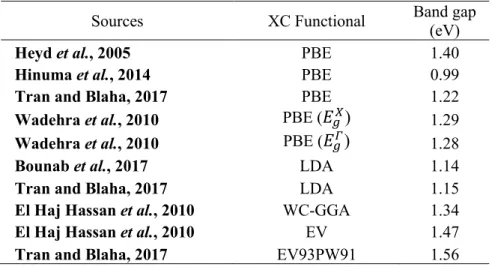
The standard deviation of the band gap curve relative to Vegard's linear trend line was measured at eV. The following expressions are derived from the polynomial curve fitting of the energy band gap values of the ternary alloy compositions, where Equation 4.1 is the calculated PBE values, Equation 4.2 is WC-GGA (El Haj Hassan et al., 2010) and Equation 4.3 is LDA ( Bounab et al., 2017). However, there is a clear indication of non-linear increase of the energy band gap values relative to the increase in the concentration of As.
There is also clear energy gap dependence on the arsenic composition, x with noticeable downward bending of the ternary alloys at 0.85.