Ramjugernath, Phase equilibria of methane clathrate hydrate from grand canonical Monte Carlo simulations, Fluid Phase Equilibria. Chapter Three (Based on Paper II): Phase Equilibria of Methane Clathrate Hydrate from Grand Canonical Monte Carlo Simulations.
CHAPTER ONE: INTRODUCTION AND BACKGROUND
- OUTLINE OF THE PRESENT STUDY
- HISTORY OF GAS HYDRATE RESEARCH
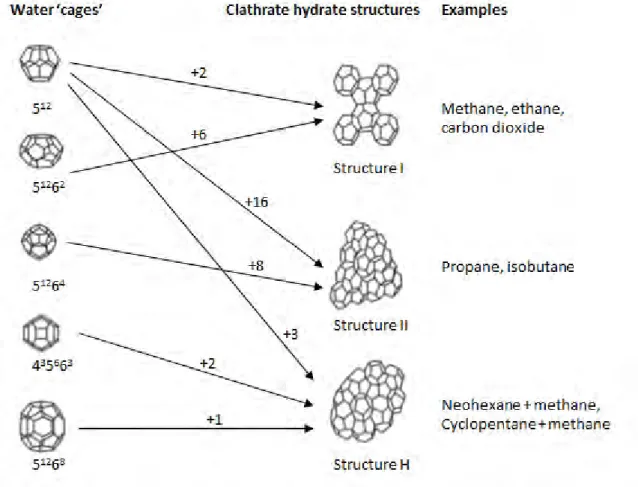
- DESCRIPTION OF GAS HYDRATE STRUCTURES
- POTENTIAL USES OF GAS HYDRATES
- MOLECULAR SIMULATIONS
- INTERMOLECULAR INTERACTIONS
- SIMULATIONS OF CLATHRATE HYDRATES IN THE LITERATURE
- BACKGROUND TO LATTICE DISTORTION THEORY
- BACKGROUND TO GCMC SIMULATIONS
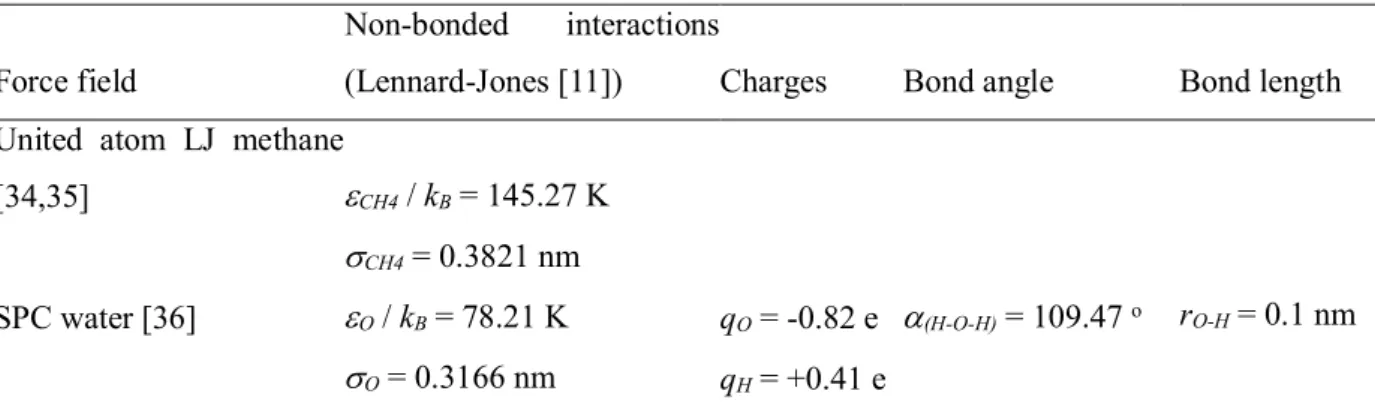
- UNLIKE LENNARD-JONES INTERACTIONS
Also in this case, the correction is applied for the different dispersion term of the LJ potential. Molecular dynamics (MD) provides information about the dynamic or time-dependent properties of the system.
CHAPTER TWO (BASED ON PAPER I)
ON THE APPLICATION OF BINARY CORRECTION FACTORS IN LATTICE DISTORTION CALCULATIONS FOR METHANE
CLATHRATE HYDRATES
INTRODUCTION
- CLATHRATE HYDRATES
- LATTICE DISTORTION (LD) THEORY
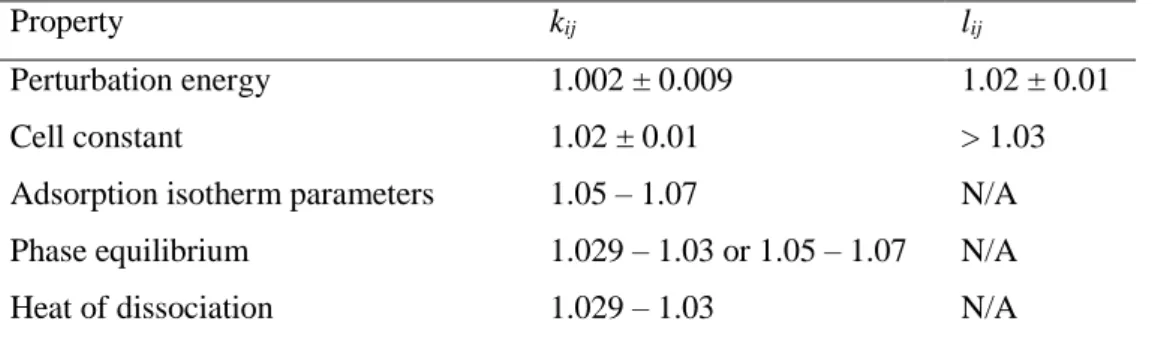
- BINARY CORRECTION FACTORS
- LD AND VAN DER WAALS-PLATTEEUW THEORY
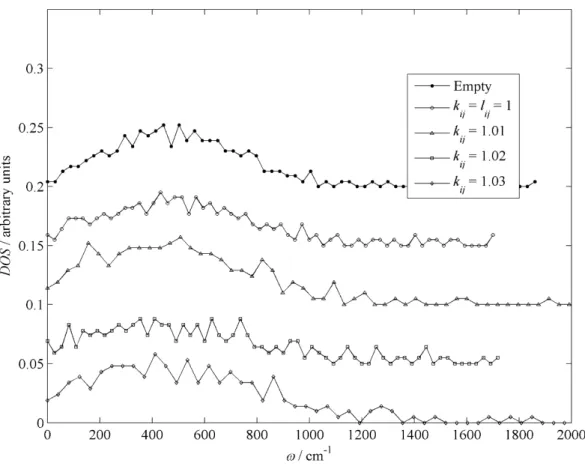
- VIBRATIONAL ENERGY
The spatial coordinates of the water molecules were then processed to calculate the vibrational contribution to the guest energy. The guest energy calculation is the sum of the static and vibrational contributions.
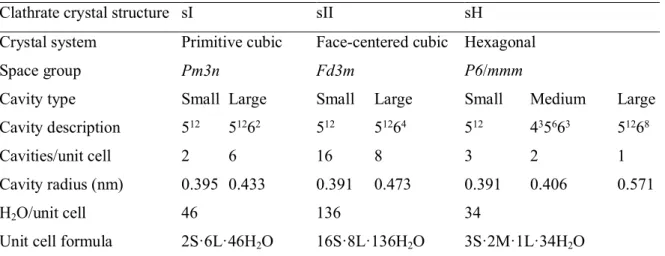
![Table 2.1. Table summarizing the crystalline structures and properties of the structure I, II, and H clathrate hydrates [2]](https://thumb-ap.123doks.com/thumbv2/pubpdfnet/10643755.0/31.892.120.736.162.415/table-summarizing-crystalline-structures-properties-structure-clathrate-hydrates.webp)
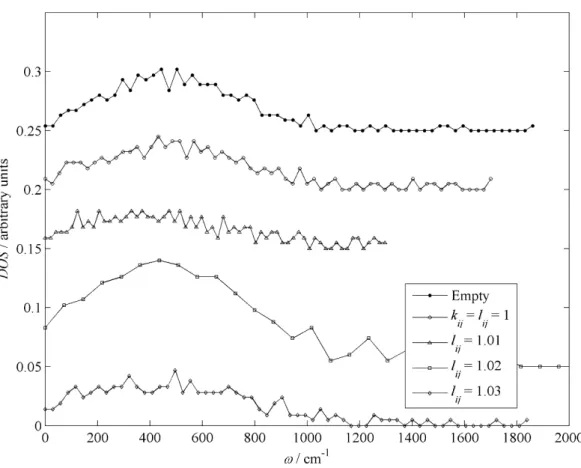
RESULTS AND DISCUSSION 1. LATTICE VIBRATIONS
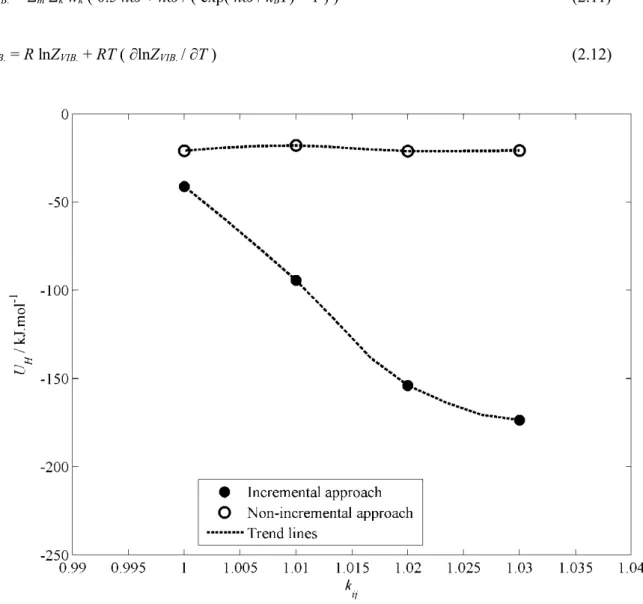
- EFFECT OF BINARY CORRECTION FACTORS ON THE PERTURBATION ENERGY
- EFFECT OF BINARY CORRECTION FACTORS ON THE CELL CONSTANT
- FORCE FIELD SENSITIVITY
- GENERAL TRENDS
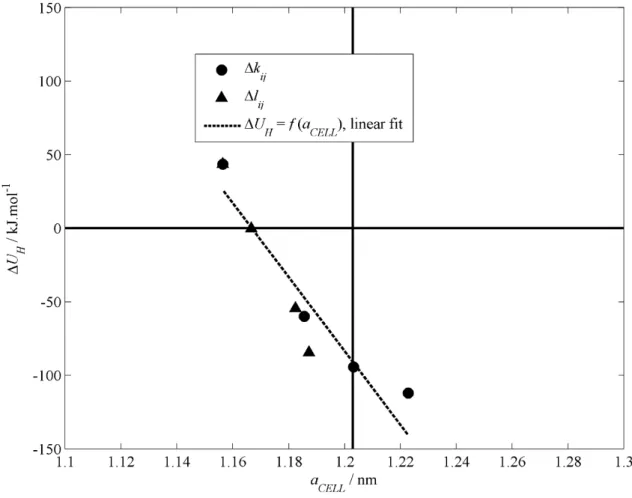
These changes are more than three orders of magnitude larger than the desired value of the perturbation energy. Another point to note is the degree of sensitivity of the perturbation energy to changes in the binary correction factors relative to each other. One reason for the lack of consistency in the determination of the binary correction factors may arise from deficiencies in the force fields used (namely SPC for water and an LJ coupled atom force field for methane in this study).
Therefore, it cannot simply be a shortcoming of the applied force fields that resulted in the sensitivity to the binary correction factors. This is because increasing the binary correction factors increases the strength of the methane-water interactions (for kij) or the equilibrium distance between the methane and water molecules (for lij). The parameters of the SPC force field are very similar to the parameters of the extended SPC (SBK/E) force field [45] as well as the TIP4P force field [46].
CONCLUSIONS
Although the trends observed in this study may be very similar to other unified atomic methane force fields, the sensitivity of ΔUH and aCELL to σTc and σPc should be considered. Thus, although ΔUH and aCELL may behave similarly as a function of kij or lij for each force field combination, the actual values of kij and lij may vary widely. An additional factor to consider when determining the general application of the results of this study is the structure-forming capacity of the various hydropower fields.
For pure water, this can be expressed as the frequency of occurrence of each geometric structure as a function of temperature. Similar quantitative results were obtained for these water force fields if a "temperature variation" was applied. Therefore, since a large number of water force fields behave in the same way when forming ordered structures, it can be concluded that the results from this study have general application, at least when an LJ gas species is used.
CHAPTER THREE (BASED ON PAPER II)
PHASE EQUILIBRIA OF METHANE CLATHRATE HYDRATE FROM GRAND CANONICAL MONTE CARLO SIMULATIONS
INTRODUCTION
This is important as details of the physical mechanism or behavior of clathrate hydrate formation or inhibition (depending on the desired application) can yield improvements in industrial processes. Moreover, purely hypothetical molecules can be investigated, providing insight into molecular behavior of clathrate hydrates. This contribution studies adsorption of methane in sI clathrate hydrate by means of GCMC simulations, as well as phase equilibria calculated from these data.
Comparisons with published results are made and the use of GCMC simulations to study phase equilibria of clathrate hydrate is demonstrated. Previous studies of adsorption in sI methane clathrate hydrate do not fully agree on the mechanism of adsorption. 16] suggested that there can be no difference between small and large types of adsorption sites in methane clathrate hydrate (for temperature ranges T < 260 K and 278 K ≤ T ≤ 328 K, respectively).
THEORY AND METHODS
- CLATHRATE HYDRATE PHASE EQUILIBRIA
- CLATHRATE HYDRATE CRYSTAL STRUCTURE
- SIMULATION DETAILS
- LANGMUIR-TYPE GAS ADSORPTION
Langmuir adsorption [44] is often used to describe the adsorption of gaseous species into the cavities of clathrate hydrate. The original form [45] of equation (3.4) also includes a term that corrects the solubility of gas species in the liquid phase. These sites are located in the centers of small and large cavities within the unit cell.
The value of the chemical potential (see Figure 3.1) was estimated in the isobaric-isothermal ensemble using the computer program "ms2" [53]. These liquid phase simulations were used to determine the chemical potential to ensure self-consistency with respect to the force fields used to describe the phases at equilibrium. In this way, any observed anomalous behavior cannot be due to inconsistencies or inconsistencies in the molecular description of the phases in equilibrium, as might be the case if, for example, an equation of state was used to determine the chemical potential of the liquid phase.
![Figure 3.1. Chemical potential (μ) of methane versus pressure (P), estimated using the isobaric isothermal ensemble with Widom’s method [53,54]](https://thumb-ap.123doks.com/thumbv2/pubpdfnet/10643755.0/57.892.133.768.122.619/figure-chemical-potential-pressure-estimated-isobaric-isothermal-ensemble.webp)
RESULTS AND DISCUSSION 1. SINGLE SITE ADSORPTION
- ADSORPTION ISOTHERMS
- PHASE EQUILIBRIA
The results of the GCMC simulations, expressed in the form of equation (3.8), are shown in Figures 3.3 to 3.5. NCH4 is the number of moles of methane adsorbed per moles of the crystal unit cell. However, the statistical uncertainties in the results of the GCMC simulations of about 5–9% and in the results of laboratory experiments of about should also be taken into account in this analysis.
The significantly larger absolute mean deviations for the fitted adsorption isotherms using equation (3.9) arise as an artifact of the temperature-dependent fitting. It was also found that phase equilibria calculated in this study using parameters derived from GCMC simulations using the SPC force field gave better predictions of the experimental data than with TIP4P/ice water. Comparison of different data sets with respect to the deviation from experimental dissociation temperature of methane clathrate hydrate.

CONCLUSIONS
Jensen, Experimental Investigation and Molecular Simulation of Gas Hydrates (Ph.D. Thesis), Technical University of Denmark, 2010. Sparks, Configurational Properties of Water Clathrates Through Molecular Simulations (Ph.D. Thesis), Massachusetts Institute of Technology, 1991 .
CHAPTER FOUR (BASED ON PAPER III)
INFLUENCE OF UNLIKE DISPERSION INTERACTIONS IN MODELING METHANE CLATHRATE HYDRATE
INTRODUCTION
Extensive discussions of the general use of combination rules can be found in the literature, although there is little work on clathrate hydrates. The use of the standard Lorentz and Berthelot rules for clathrate hydrate systems has only recently been discussed in the literature [18]. A comprehensive study was recently carried out to determine the effects of the Lorentz and Berthelot combination rules on adsorption in argon clathrate hydrates [6].
Changes in the Lorentz and Berthelot combination rules resulted in significant changes in the adsorption for the sII and sH clathrate hydrate structures, due to the difference in size between the cages present in these structures. This is achieved in a computationally convenient way [18] by introducing a correction factor into the Berthelot rule. The contribution to the polarizability is purely a dispersion/energetic effect, and thus the size parameter remains unaffected.
THEORY AND METHODS
- CLATHRATE HYDRATE PHASE EQUILIBRIA
- INTERMOLECULAR INTERACTIONS
- GRAND CANONICAL MONTE CARLO SIMULATIONS
- DIRECT ESTIMATION OF THE HEAT OF DISSOCIATION
Thus, phase equilibria are inferred using the dependence of occupancy on pressure and temperature. In the case of using GCMC simulations (the first approach discussed in this study), the heat of dissociation can only be inferred by examining the effect of the adsorption isotherm parameters on the slope of the dissociation pressure curve. In this work, the adsorption isotherms and dissociation heat of methane clathrate hydrate were directly studied for different values of the well depth LJ, ij.
The static contribution to the internal energy was calculated from the force field using the spatial coordinates of the interaction sites. A similar procedure was used for the liquid phases as for estimating the chemical potential of methane gas as described in Section 4.2.3. The heat of dissociation was determined from the enthalpy differences of the combined hydrate phase, methane gas, and liquid water [59].
RESULTS AND DISCUSSION
- GRAND CANONICAL MONTE CARLO SIMULATIONS
- DIRECT ESTIMATION OF THE HEAT OF DISSOCIATION
The adsorption isotherm parameters A and B (see Eq. 4.4)), adapted to the results of the GCMC simulations, are shown in Table 4.2. Since the slope of the dissociation pressure curve is related to the heat of dissociation (see Eq. 4.5)), any effects of unequal dispersion interactions can also be investigated via Fig. Gibbs free energy of the sI-methane clathrate hydrate relative to the reference case (ie kij = 1) for different kij values, at T = 280 K and P = 10 MPa.
There is a monotonic trend for the heat of dissociation in terms of the uneven distribution, suggesting that fit to experimental data can be achieved. The decrease in the heat of dissociation with increasing kij suggests that increasing the unequal dispersion interaction reduces the enthalpy difference between the solid phase and the weighted sum of the liquid phases (gaseous methane and liquid water). Therefore, if the crystal lattice calculations are sensitive to changes in the dissimilar interactions, this sensitivity can carry over into the estimate of the dissociation enthalpy.
CONCLUSIONS
A major drawback of directly estimating the heat of dissociation, as opposed to using GCMC simulations to derive phase equilibria of clathrate hydrates, is that the coating behavior cannot be calculated. At best, Langmuir adsorption isotherm parameters can be fitted to the phase equilibria estimated via the Clausius–Clapeyron equation, although no details of molecular behavior can be determined by this approach. To fully study various aspects of clathrate hydrate systems by computational means, it is necessary to employ a variety of methods.
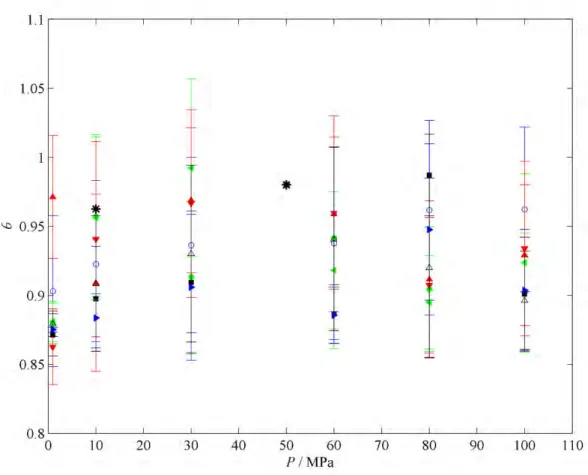
The different dispersion interaction monotonically affects the calculated dissociation heats, as well as the phase equilibria estimated via the Clausius-Clapeyron equation. Plot of overall fractional occupancy θ (see Equation (4.6)) versus pressure P for sI methane clathrate hydrate at T = 280 K. Plot of overall fractional occupancy θ (see Equation (4.6)) versus pressure P for sI methane clathrate hydrate at T = 300 K.
CHAPTER FIVE
GENERAL DISCUSSION AND CONCLUSIONS
- SUMMARY OF THESIS FINDINGS
- UNLIKE INTERACTIONS AND FITTING TO PROPERTIES
- SYSTEM RESPONSES TO CHANGING UNLIKE INTERACTIONS
- OUTLOOK AND FUTURE WORK
After examining the solution space for methane clathrate hydrate described by these two parameters, it was clear that the difference in It can be noted that the detailed study of the effect of changing different interactions on adsorption on methane clathrate hydrate was not demonstrated before. The use of a flexible water network in GCMC simulations should be considered in the context of the statistical mechanical theory used to calculate phase equilibria [6].
Regarding the sensitivity of the approaches considered in this thesis for parameterizing force fields, lattice deformation calculations and GCMC simulations yield different results. Thus, increasing the strength of the non-uniform dispersion interaction results in a more negative disruption energy and an increase in the cell constant. This is due to the fact that when adjusting these parameters to the results of the GCMC simulations, aggregated data sets are taken into account.
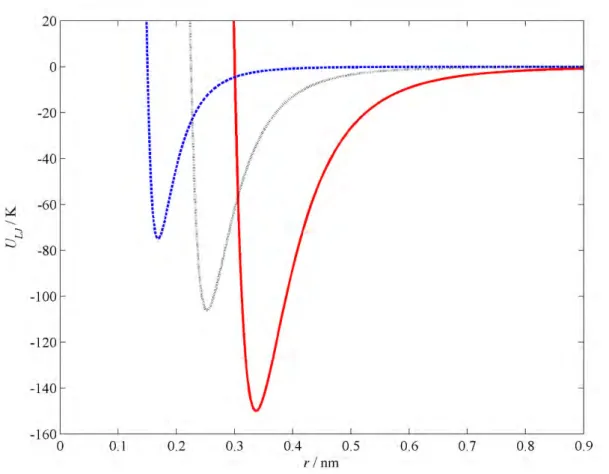

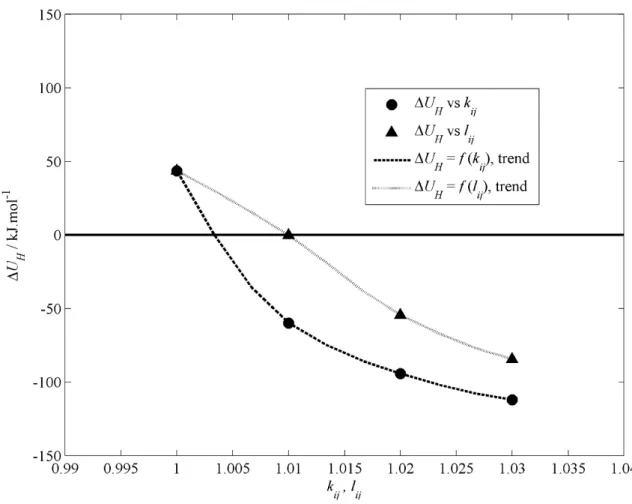
![Figure 2.5. Effect of k ij and l ij on a CELL . The solid horizontal line is the experimental value of a CELL (1.203 nm) [15]](https://thumb-ap.123doks.com/thumbv2/pubpdfnet/10643755.0/42.892.151.769.139.641/figure-effect-cell-solid-horizontal-experimental-value-cell.webp)