PENGELOMPOKAN FRAGMEN METAGENOM DENGAN
METODE GROWING SELF ORGANIZING MAP
MARLINDA VASTY OVERBEEK
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI TESIS DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa tesis berjudul Pengelompokan Fragmen Metagenom dengan Metode Growing Self Organizing Map adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir tesis ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
RINGKASAN
MARLINDA VASTY OVERBEEK. Pengelompokan Fragmen Metagenom dengan Metode Growing Self Organizing Map. Dibimbing oleh WISNU ANANTA KUSUMA dan AGUS BUONO.
Metagenom adalah penelitian tentang bagaimana menganalisis mikrob berskala besar dan memperbolehkan adanya pengkulturan secara langsung. Pengelompokan fragmen metagenom secara langsung bisa berakibat fatal karena bisa menyebabkan terjadinya interspesies chimeras atau kesalahan dalam perakitan fragmen metagenom. Pengelompokan fragmen metagenom pada lingkungan juga pada umumnya menggunakan supervised learning, sedangkan supervised learning merupakan pembelajaran yang menggunakan contoh dan bergantung pada ketersediaan data latih. Selain itu, pengelompokan juga menggunakan panjang fragmen yang panjang, yaitu ≥ 8 kbp dan berkomunitas kecil atau kurang dari 100 mikrob. Tujuan penelitian ini adalah untk menganalisis efektifitas dan efisiensi metode Growing Self Organizing Map dalam pengelompokan mikrob yang berskala besar dengan panjang fragmen yang pendek berdasarkan frekuensi oligonukleotida. Frekuensi oligonukleotida yang digunakan adalah trinukleotida, tetranukleotida, dan juga kombinasi frekuensi yang memperhatikan kondisi don’t care, yaitu spaced k-mer. Untuk ekstraksi fitur, digunakan k-mer frequency dan spaced k-mer frequency.
Berdasarkan uji kombinasi parameter menggunakan frekuensi oligonukleotida, kombinasi terbaik antara Learning Rate dan Neighborhood Size untuk frekuensi trinukleotida adalah 0.1 untuk Learning Rate, 1 untuk Neighborhood Size dengan perhitungan quantization error adalah 0.531, 0.101 untuk topographic error, dan 16.84% untuk persentase error. Kombinasi terbaik tetranukleotida adalah 0.75 untuk Learning Rate dan 1 untuk Neighborhood Size, dengan memberikan nilai error 0.886 untuk quantization error, 0.09 untuk topographic error, dan 15.43% untuk persentase error. Untuk spaced k-mer, kombinasi terbaik adalah 0.5 untuk Learning Rate dan 1 untuk Neighborhood Size dengan quantization error adalah 0.665, 0.06 untuk topographic error dan 13.07% untuk persentase error. Perhitungan kombinasi untuk ketiga frekuensi oligonukleotida menggunakan map size dan dan training lenght yang sama, yaitu [10 10] dan 10 epochs.
Dari hasil kombinasi parameter, frekuensi spaced k-mer menjadi frekuensi terbaik untuk pengelompokan fragmen metagenom dengan metode Growing Self Organizing Map. Dengan menggunakan map size yang berukuran antara [100 – 500], unit peta dari 100 – 5000 unit, dan training lenght 10 epochs, didapatkan hasil terbaik pelatihan adalah pada map size [100 150] dengan unit peta sebanyak 300 unit. Waktu latih yang diperlukan adalah 51 menit dengan persentase error 6.43%.
SUMMARY
MARLINDA VASTY OVERBEEK. Clustering Metagenome Fragments using Growing Self Organizing Map. Supervised by WISNU ANANTA KUSUMA and AGUS BUONO.
Metagenome is a research about analyzing microbes in the large community and allowed the culture-independent. The microorganism samples taken directly from environment is not easy to assembly because contains mixture microorganism. If sample complexity is very high and come from high diversity environment, difficulties of assembling DNA sequence are increasing because the interspecies chimeras can be happen. Clustering commonly using supervised learning, but the supervised learning depends on avaibillity of data training. Because of that, in this research we used unsupervised learning to clustering the metagenome fragments. Beside that, clustering usually using the longer
fragments, which is ≥ 8 kbp and have a small community (less than 100
microorganism). The purpose of this research is to analyze the effectiveness and efficiency of Growing Self Organizing Map to the clustered large community of metagenome fragments. We used trinucleotide, tetranucleotide, and combination
of oligonucleotide frequency that consider the don’t care situation called spaced k -mer frequency as a features. As a feature extraction, we using -mer and spaced k-mer.
Based on parameter combination using oligonucleotide frequency, the best combine between Learning Rate and Neighborhood Size is a spaced k-mer frequency. We tested to get a better parameter combinatoin into [10 10] map size and 10 epochs training lenght. Error to mapped metagenome fragments using spaced k-mer frequency is 0.665 for quantization error, 0.06 for topographic error and 13.07% for error percentage.
© Hak Cipta Milik IPB, Tahun 2013
Hak Cipta Dilindungi Undang-Undang
Dilarang mengutip sebagian atau seluruh karya tulis ini tanpa mencantumkan atau menyebutkan sumbernya. Pengutipan hanya untuk kepentingan pendidikan, penelitian, penulisan karya ilmiah, penyusunan laporan, penulisan kritik, atau tinjauan suatu masalah; dan pengutipan tersebut tidak merugikan kepentingan IPB
Tesis
sebagai salah satu syarat untuk memperoleh gelar Magister Komputer
pada
Program Studi Ilmu Komputer
PENGELOMPOKAN FRAGMEN METAGENOM DENGAN
METODE GROWING SELF ORGANIZING MAP
SEKOLAH PASCASARJANA INSTITUT PERTANIAN BOGOR
BOGOR 2013
PRAKATA
Puji dan syukur penulis panjatkan kepada Tuhan yang Maha Kuasa atas segala karunia-Nya sehingga karya ilmiah ini berhasil diselesaikan. Penelitian ini sudah dikerjakan dari bulan September 2012 dengan judul Pengelompokan Fragmen Metagenom dengan Metode Growing Self Organizing Map.
Terima kasih penulis ucapkan kepada Bapak Dr Eng Wisnu Ananta Kusuma, ST, MT dan Bapak Dr Ir Agus Buono, MSi, MKom selaku pembimbing yang telah banyak memberi saran, kepada Bapak Dr Ir Iman Rusmana, MSi selaku penguji. Selain itu, penulis menyampaikan terima kasih kepada semua dosen dan staf Departemen Ilmu Komputer IPB yang telah membantu selama proses penelitian. Ungkapan terima kasih juga disampaikan kepada Papa John dan Mama Naniek, Mbak Yoanita, Kak Alex, Mas Andrew, Mbak Santhy, Kevin Joshua, Mama Yosina, dan Fajar Ndolu atas doa, perhatian dan kasih sayangnya. Teman-teman Dwi Regina (Frinsa, Mentari, Inna, Toyibah, Astrid, Lian, Erlisa), teman sepembimbingan Bapak Wisnu (Dian, Aa Bahrul, Kang Asril) dan teman-teman seperjuangan angkatan 13 Ilmu Komputer IPB yang selalu bersama penulis dua tahun ini, terima kasih atas dukungannya. Penulis juga tidak lupa berterima kasih pada jajaran dosen dan staf STIKOM Uyelindo Kupang atas semua bantuan yang diberikan kepada penulis.
Semoga karya ilmiah ini bermanfaat.
DAFTAR ISI
DAFTAR TABEL vi
DAFTAR GAMBAR vi
DAFTAR LAMPIRAN vi
1 PENDAHULUAN 1
Latar Belakang 1
Perumusan Masalah 3
Tujuan Penelitian 3
Manfaat Penelitian 3
Ruang Lingkup Penelitian 3
2 TINJAUAN PUSTAKA 4
Metagenom 4
Ekstraksi Ciri 4
Growing Self Organizing Map 5
3 METODE PENELITIAN 8
4 HASIL DAN PEMBAHASAN 17
5 SIMPULAN 36
DAFTAR PUSTAKA 37
LAMPIRAN 40
DAFTAR TABEL
1 Dimensi hasil ekstraksi ciri 10
2 Filum berdasarkan NCBI Taxonomy Browser 11
3 Pembagian mikrob data latih dan data uji 19
4 Pembangkitan data latih 19
5 Pembangkitan data uji 20
6 Perhitungan quantization error pada trinukleotida 24 7 Perhitungan topographic error pada trinukleotida 24 8 Perhitungan persentase error pada trinukleotida 24 9 Perhitungan quantization error pada tetranukleotida 26 10 Perhitungan topographic error pada tetranukleotida 26 11 Perhitungan persentase error pada tetranukleotida 27 12 Perhitungan quantization error pada spaced k-mer 29 13 Perhitungan topographic error pada spaced k-mer 29 14 Perhitungan persentase error pada spaced k-mer 29
15 Parameter pengujian 32
16 Hasil pelatihan frekuensi spaced k-mer 32
17 Daftar organisme yang memiliki kesamaan dari hasil alignment
Bacteroides fragilis 638R pada BLAST 34
DAFTAR GAMBAR
1 Binning sampel metagenomik (Kusuma 2012) 4
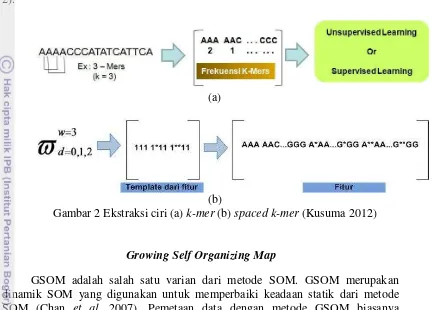
2 Ekstraksi ciri (a) k-mer (b) spaced k-mer (Kusuma 2012) 5
3 Aturan inisialisasi node (Zhu dan Zhu 2010) 7
4 Skema penelitian pengelompokan fragmen metagenom 8
5 Prosedur analisis 9
6 Contoh hasil simulasi MetaSim 10
7 Praproses data dengan decimal scaling 11
8 Blok diagram pengelompokan dengan GSOM 12
9 Fase inisialisasi 13
10 Inisialisasi starting node 13
11 Best Matching Unit (Vesanto et al. 2000) 15
12 Pengukuran quantization error 15
13 Pengukuran topographic error 16
14 Matriks komposisi salah satu frekuensi oligonukleotida 17 15 Matriks decimal scaling salah satu frekuensi oligonukleotida 18 16 Inisialisasi bobot pada frekuensi oligonukleotida 21
17 Pemetaan frekuensi trinukleotida 22
18 Pemetaan frekuensi tetranukleotida 22
19 Pemetaan frekuensi spaced k-mer 23
26 Perhitungan quantization error pada spaced k-mer 30 27 Perhitungan topographic error pada spaced k-mer 30 28 Perhitungan persentase error pada spaced k-mer 31 29 Hasil pengelompokan Bacteroides fragilis 638R dengan 1024 reads 33 30 Data yang digunakan dengan panjang sekuens query 1000 (1 kbp) 33 31 Hit dari 17 organisme yang memiliki kesamaan dengan Bacteroides
fragilis 638R 34
DAFTAR LAMPIRAN
1 Daftar mikrob yang digunakan sebagai data latih 40
2 Daftar mikrob yang digunakan sebagai data uji 45
3 Hasil analisis pengelompokan frekuensi trinukleotida map size [10 10] dengan Learning Rate 0.1 dan Neighborhood Size 1 48 4 Hasil analisis pengelompokan frekuensi tetranukleotida map size [10
10] dengan Learning Rate 0.75 dan Neighborhood Size 1 51 5 Hasil analisis pengelompokan frekuensi spaced k-mer map size [10 10]
dengan Learning Rate 0.5 dan Neighborhood Size 1 54 6 Pohon taksonomi BLAST dari organisme yang memiliki kesamaan
1
1 PENDAHULUAN
Latar Belakang
Penelitian tentang metagenom terus berkembang dalam lingkup biologi molekuler. Analisis tentang metagenom disebut dengan metagenomik, yaitu analisis tentang mikrob yang berskala besar yang diambil langsung dari habitat asal mikrob tersebut (Chan et al. 2007; O’Malley 2012). Pengisolasian mikrob secara langsung seringkali memiliki kendala untuk mengetahui komunitas sesungguhnya dari suatu ekosistem karena hanya 1% mikrob yang dapat diisolasi langsung dari lingkungan (Harayama et al. 2004). Contoh dari kesulitan untuk isolasi lagsung dari lingkungan adalah proyek laut Sargasso (Venter et al. 2004). Istilah low-abundance digunakan untuk menggambarkan keadaan ini. Low-abundance adalah rendahnya representasi relatif keanekaragaman mikrob dalam sampel lingkungan sehingga masih banyak mikrob yang belum dikenali dan dimanfaatkan (Chan et al. 2007; Harayama et al. 2004). Low-abundance pada fragmen metagenom yang berukuran besar sering menimbulkan kendala dalam perakitan genom dan menyebabkan mikrob sulit dikelompokan secara filogenetik (Chan et al. 2007). Kesalahan dalam perakitan fragmen metagenom disebut interspecies chimeras (Meyerdierks dan Glockner 2012).
Untuk menyelesaikan permasalahan tersebut, binning digunakan untuk mengelompokan mikrob berdasarkan tingkatan taksonomi. Ada dua pendekatan binning, yaitu berdasarkan homologi dan berdasarkan komposisi. Binning berdasarkan homologi melakukan pencarian penjajaran sekuens dengan membandingkan fragmen metagenom dengan basis data sekuens antara lain National Centre for Biotechnology Information (NCBI) dan hasilnya akan disimpulkan pada tiap level taksonomi. Hal tersebut menyebabkan pendekatan dengan homologi membutuhkan banyak waktu dalam proses pengelompokan. Contoh metode yang menggunakan pendekatan homologi adalah BLAST (Wu 2008; Zheng dan Wu 2009) dan MEGAN (Huson et al. 2007).
2
Sebagian besar proses binning masih menggunakan pembelajaran dengan contoh (supervised learning). Pembelajaran dengan contoh bergantung pada ketersediaan data latih padahal data latih yang tersedia tidak cukup merepresentasikan keragaman mikrob (Prabhakara dan Acharya 2012). Pembelajaran dengan observasi (unsupervised learning) memberikan solusi terhadap keterbatasan data latih yang tersedia karena unsupervised learning akan menyusun data fragmen metagenom secara lebih terstruktur sebelum perbandingan sekuens dilakukan. Dengan demikian fragmen metagenom akan lebih cepat dan lebih kuat (robust) untuk dirakit (Nasser et al. 2008).
Dari beberapa pendekatan binning berdasarkan komposisi dengan unsupervised learning, metode GSOM memberikan hasil terbaik dalam pemetaan fragmen metagenom. Oleh sebab itu, pada penelitian tentang pengelompokan fragmen metagenom ini akan menggunakan metode GSOM.
Metode GSOM merupakan perbaikan dari keadaan statik metode Kohonen SOM (Chan et al. 2007). GSOM sukses memetakan data dalam bentuk microarray (Hsu et al. 2003) dan juga memetakan data prokariota dengan panjang
≥ 8 kbp (Chan et al. 2007). Hasil yang didapatkan adalah pada pengelompokan mikrob dengan empat frekuensi oligonukleotida (di-, tri-, tetra-, dan pentanukleotida) pada tiga dataset mikrob, pengelompokan menggunakan frekuensi dinukleotida tidak terlalu memberikan hasil yang baik sehingga disarankan utuk menggunakan frekuensi oligonukleotida yang lebih tinggi. Dalam perbandingan kecepatan, GSOM mengalami peningkatan kecepatan 37 % dibandingkan metode SOM pada dua dataset pertama dan untuk keseluruhan tiga dataset terjadi peningkatan kecepatan 7 % - 15 %.
Penelitian fragmen metagenom menggunakan unsupervised learning umumnya hanya menggunakan komunitas yang kecil. Sedangkan untuk ekstraksi ciri, pengelompokan fragmen metagenom masih menggunakan k-mer dan belum memperhatikan kondisi don’t care. Ekstraksi ciri dengan memperhatikan kondisi don’t care disebut dengan spaced k-mer (Kusuma 2012). Spaced k-mer menyediakan vektor berdimensi lebih kecil yang berisi informasi yang lebih kaya dan berguna dibandingkan dengan vektor masukan hasil ekstraksi fitur menggunakan k-mer (Kusuma 2012).
Pada penelitian ini digunakan komunitas spesies yang cukup besar, yaitu 300 spesies dan data spesies tersebut diambil dari basis data NCBI. Panjang fragmen yang digunakan adalah 1 kbp dengan frekuensi oligonukleotida trinukleotida dan tetranukleotida. Alasan digunakan fragmen yang pendek karena pada penelitan terdahulu, panjang fragmen yang digunakan adalah fragmen yang
3
Perumusan Masalah
Adapun permasalahan yang akan menjadi bahan analisis adalah mengetahui berapa tingkat akurasi efektifitas dan efisiensi menggunakan metode GSOM menggunakan frekuensi trinukleotida, tetranukleotida, dan spaced k-mer pada panjang fragmen yang pendek.
Tujuan Penelitian
Menganalisis efektifitas dan efisiensi metode GSOM dalam pengelompokan mikrob berskala besar pada tingkat taksonomi filum berdasarkan frekuensi trinukleotida, tetranukleotida dan spaced k-mer dengan fragmen yang pendek (1 kbp).
Manfaat Penelitian
Adapun manfaat dari penelitian yang dilakukan adalah untuk memberikan landasan bagi penelitian lanjutan di bidang metagenomik, khususnya yang memerlukan informasi kekerabatan antar organisme yang terdapat pada komunitas atau sampel yang diamati.
Ruang Lingkup Penelitian
Ruang lingkup penelitian ini adalah :
1. Data latih terdiri atas 200 mikrob yang berasal dari 20 filum
2. Data uji yang terdiri atas 100 mikrob yang termasuk dalam taksonomi yang sama dengan data latih untuk mengetahui kualitas kebaikan pengelompokan fragmen metagenom dengan GSOM
4
2 TINJAUAN PUSTAKA
Metagenom
Metagenomik adalah penelitian tentang mikrob yang sampelnya diambil secara langsung dari komunitas mikrob. Umumnya komunitas mikrob tersebut memiliki keanekaragaman yang tinggi dan berskala besar (Chan et al. 2007;
O’Malley 2012).
Pengambilan sampel langsung dari lingkungan atau isolasi secara langsung sering menyebabkan terjadinya masalah. Masalah yang sering muncul adalah ketika sampel yang diambil memiliki kompleksitas yang tinggi, yaitu setiap mikrob yang berada dalam sampel memiliki kekerabatan yang dekat dan hal tersebut sering menyebabkan kesalahan dalam perakitan fragmen metagenom yang disebut dengan interspecies chimeras (Meyerdierks dan Glockner 2010).
Untuk menghindari terjadinya interspecies chimeras, maka fragmen metagenom perlu dikelompokan berdasarkan tingkat taksonomi atau disebut dengan binning (Meyerdierks dan Glockner 2010). Pada pengelompokan atau binning fragmen metagenom, sangat mungkin tiap kelompok atau bin memiliki mikrob yang sama berdasarkan tingkat taksonominya. Gambar 1 menunjukkan bagaimana binning fragmen metagenom dan proses perakitan DNA diperlukan di dalam proses analisis metagenom (Kusuma 2012).
Gambar 1 Binning sampel metagenomik (Kusuma 2012)
Ekstraksi Ciri
K-mer adalah substring dengan panjang k (k adalah panjang fragmen metagenom). Analisis dari k-mer digunakan untuk menemukan frekuensi dari semua k-mer. Pola kemunculan k adalah pola yang menampilkan k pada suatu waktu dalam suatu sekuens (Choi dan Cho 2002).
Pola kemunculan dalam dalam sekuens dihitung menggunakan empat basa utama (A, T, G, dan C) dipangkat dengan rangkaian pasangan basa yang ingin digunakan (pola kemunculan : , dengan ). Selain menggunakan frekuensi k-mer, digunakan spaced k-mer yang memperhitungkan kondisi don’t care.
Spaced k-mer dikemukakan oleh Kusuma (2012) yang menyimpulkan bahwa terbaik dari klasifikasi metagenom dicapai dengan menggunakan ,
5 adalah posisi dari kondisi don’t care (*). Dari hasil percobaan, didapatkan hasil akurasi terbaik adalah pada pola 111 1*11 1**11. Hasil dari perhitungan ekstraksi fitur menggunakan frekuensi k-mer dan spaced k-mer ini yang akan digunakan sebagai masukkan pada unsupervised learning dan supervised learning (Gambar 2).
(a)
(b)
Gambar 2 Ekstraksi ciri (a) k-mer (b) spaced k-mer (Kusuma 2012)
Growing Self Organizing Map
GSOM adalah salah satu varian dari metode SOM. GSOM merupakan dinamik SOM yang digunakan untuk memperbaiki keadaan statik dari metode SOM (Chan et al. 2007). Pemetaan data dengan metode GSOM biasanya merupakan data yang berdimensi tinggi. Hasil pemetaan ditampilkan berdasarkan topologi data, jadi data yang mirip akan dipetakan berdasarkan kedekatan ciri atau karakteristiknya pada peta dua dimensi atau tiga dimensi.
GSOM memiliki tiga fase utama, yaitu fase inisialisasi, fase growing, dan fase smoothing. Langkah algoritma GSOM adalah sebagai berikut (De Silva et al. 2007; Zhu dan Zhu 2010) :
1. Fase Inisialisasi
Inisialisasi bobot vektor dan awal node (biasanya empat node) dengan angka random antara 0 dan 1.
Hitung Growth Threshold (GT) dari dimensi dataset D berdasarkan nilai Spread Factor(SF) menggunakan formula :
(1) 2. Fase Growing
a) Tentukan node masukkan pada jaringan
b) Tentukan bobot vektor yang berdekatan dengan vektor masukkan yang dipetakan sebagai winner, gunakan jarak Euclidean untuk mengukur. Langkah ini dapat disimpulkan dengan menentukan dimana
6
vektor, adalah posisi vektor untuk node-node dan adalah himpunan dari angka natural.
c) Sesuaikan bobot vektor yang diaplikasikan hanya kepada tetangga dari winner dan winner itu sendiri. Tetangga adalah neuron disekitar winner, tapi pada GSOM, tetangga awal diseleksi berdasarkan kesesuaian bobot yang kecil, berbeda dengan SOM yang berdasarkan penyesuaian bobot lokal. Besar dari penyesuaian (laju pembelajaran) direduksi secara eksponensial sejalan dengan iterasi yang terjadi. Meskipun tetangga, bobot yang berdekatan dengan winner lebih mudah disesuaikan dibandingkan yang jaraknya jauh. Penyesuaian bobot dideskripsikan sebagai berikut : pada nilai pada node yang terdapat di peta saat waktu ke .
Untuk nilai fungsi tetangga digunakan fungsi Gaussian. Formulasi pengukuran ukuran node tetangga adalah sebagai berikut :
Dengan adalah jarak antara dan dengan sebagai parameter
‘lebar efektif’ dari lingkungan.
d) Naikkan nilai error pada winner (nilai error adalah perbedaan antara vektor masukkan dan bobot vektor).
e) Ketika dengan adalah total error pada node dan adalah Growth Threshold. Node akan berubah jika adalah node batas. Distribusi bobot pada tetangga jika bukan merupakan node batas. f) Inisialisasi bobot node baru (Gambar 3) dengan mengikuti beberapa lama juga boleh memiliki tetangga yang tidak bertetangga secara bersebrangan dengan node yang baru. Aturan ini mirip dengna (i), tetapi memiliki perbedaan pada posisi tetangga. Ketika kedua kondisi dipenuhi, gunakan aturan (i)
7 (iii) Berada antara dua node yang lama. Jika di definisikan maka :
(5) (iv) Memiliki satu saja tetangga node yang lama. Jika di definisikan
maka :
Gambar 3 Aturan inisialisasi node (Zhu dan Zhu 2010) g) Inisialisasi laju pembelajaran pada bobot node yang baru
h) Ulangi langkah b sampai g hingga semua masukkan di representasikan dan growth node atau node yang mengalami perubahan mencapai level maksimum
3. Fase Smooting
a) Untuk setiap node pada peta, akan terbentuk set yang terdiri dari semua item masukkan dimana item masukkan tersebut adalah item yang di referensikan pada node dan memiliki jarak terdekat
b) Untuk setiap node pada peta, ambil satu item yang di referensikan dan yang merupakan rata-rata dari daftar gabungan topologi set tetangga (N : angka natural) node tersebut.pada tahap ini Learning Rate dan Neighborhood Size akan di redukasi. Rata-rata jika di definisikan adalah sebagai berikut :
c) Ulangi langkah a dan b sampai memperoleh peta yang stabil
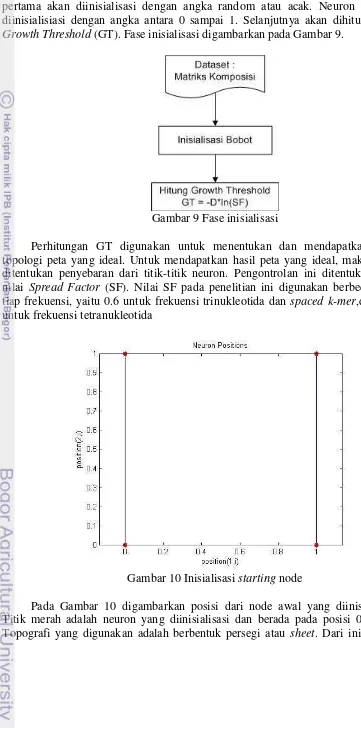
Growth Threshold , berdasarkan dimensi dataset dan Spread Factor . adalah penentu awal nilai dengan rentang 0 sampai 1, 0 adalah nilai paling mungkin untuk menyebar dan 1 adalah penyebaran maksimum. Batasan penyebaran dengan nilai terkecil adalah nilai pemetaan awal yang ideal. Sekali pengelompokan yang signifikan teridentifikasi, maka bisa dijadikan sebagai pijakan analisis selanjutnya dengan nilai yang tinggi.
6)
8
3 METODE PENELITIAN
Penelitian ini menggunakan data fragmen metagenom dari 300 mikrob dan kemudian dikelompokan berdasarkan tingkat taksonomi filum. Teknik pengambilan data fragmen metagenom yang digunakan adalah cluster sampling. Teknik cluster sampling adalah teknik yang menggunakan sampel yang memiliki jumlah item yang banyak pada suatu kelompok atau koleksi dan merupakan teknik yang sederhana serta rendah biaya (Scheafffer et al. 1990).
Sesuai dengan tujuan penelitian ini, metode GSOM digunakan untuk pengelompokan fragmen metagenom. Data awal akan disimulasi menggunakan MetaSim (Richter et al. 2008) dan menghasilkan sekuens DNA. Hasil simulasi ini yang akan digunakan pada pengekstraksian ciri sehingga didapat matriks komposisinya. Selanjutnya fragmen metagenom akan dikelompokan menjadi 20 kelompok yang berbeda berdasarkan kesamaan dari pemetaan yang dihasilkan. Ilustrasi pemetaan fragmen metagenom, ditunjukkan pada Gambar 4.
9
Gambar 5 Prosedur penelitian
Data Penelitian
10
Gambar 6 Contoh data hasil simulasi MetaSim.
Ekstraksi Ciri
Ekstraksi ciri adalah pembacaan frekuensi oligonukleotida (trinukleotida dan tetranukleotida) dengan k-mer dan juga ekstraksi menggunakan spaced k-mer yang memperhatikan kondisi don’t care pada perhitungan frekuensi oligonukleotida. Ekstraksi ciri akan menampilkan pola kemunculan k pada suatu waktu dalam suatu sekuens. Pada penelitian ini, dimensi hasil ekstraksi ciri adalah sebagai berikut seperti pada Tabel 1 :
Tabel 1 Dimensi hasil ekstraksi ciri
Frekuensi Oligonukleotida Dimensi
Trinukleotida 64 × total fragmen
Tetranukleotida 256 × total fragmen
Spaced k-mer 192× total fragmen
Praproses Data
Untuk mencegah adanya hasil implementasi yang bias, maka pengelompokan fragmen metagenom didahului dengan normalisasi data hasil ekstraksi fitur. Normalisasi data adalah salah satu bagian dari data transformasi, yaitu teknik mengubah data menjadi nilai yang lebih mudah untuk dipahami (Han et al. 2012).
Tujuan lebih khusus dari normalisasi data adalah mendapatkan bobot yang sama dari semua atribut data dan tidak bervariasi atau hasil dari pembobotan tersebut tidak terdapat atribut yang lebih prior atau dianggap lebih utama dari pada yang lain.
Untuk penelitian ini, normalisasi data yang digunakan adalah decimal scaling. Data fragmen metagenom akan diubah bobotnya menjadi data yang memiliki rentang [0, 1] menggunakan transformasi linear sederhana (Vesanto et al. 2000). Contoh hasil praproses data pada Gambar 7.
dengan adalah dataset.
(8)
>r1.1 |SOURCES={GI=298489614,bw,4206109-4207109}| ERRORS={}
11
Gambar 7 Praproses data dengan decimal scaling Pembagian Data Latih dan Data Uji
Jumlah data adalah 200 mikrob untuk data latih dengan total jumlah fragmen yang digunakan adalah 200 000 fragmen. Sedangkan untuk data uji digunakan 100 mikrob dengan total jumlah fragmen sebanyak 100 000 fragmen. Perkiraan fragmen per mikrob adalah sebanyak 1000 fragmen. Frekuensi oligonukleotida yang digunakan juga beragam untuk masing-masing dataset, yaitu trinukleotida, tetranukleotida, dan juga menggunakan spaced k-mer.
Pengelompokan dengan Growing Self Organizing Map
Pengelompokan dilakukan menggunakan matriks komposisi hasil ekstraksi fitur. Normalisasi dilakukan pada matriks komposisi agar perhitungan tidak menghasilkan data yang terlalu bervariasi dan bobot yang sama. Data kemudian akan dikelompokan berdasarkan tingkat filum sebanyak 20 kelompok sesuai dengan NCBI Taxonomy Browser (Federhen 2012). Filum yang digunakan berada pada Tabel 2.
Tabel 2 Filum berdasarkan NCBI Taxonomy Browser
No Filum
1 Actinobacteria (high G+C gram positive bacteria)
2 Aquificae
3 Bacteroidetes
4 Chlorobi
5 Chlamydiae
6 Verrucomicrobia
7 Chloroflexi (green non sulfur bacteria)
8 Cyanobacteria (blue green algae)
9 Deinococcus-thermus
10 Acidobacteria
11 Firmicutes (gram positive bacteria)
12 Fusobacteria
13 Gemmatimonadetes
14 Nitrospirae
15 Planctomycetes
16 Proteobacteria (purple bacteria and relative)
17 Spirochaetes
18 Synergistetes
19 Tenericutes
20 Thermotogae
12
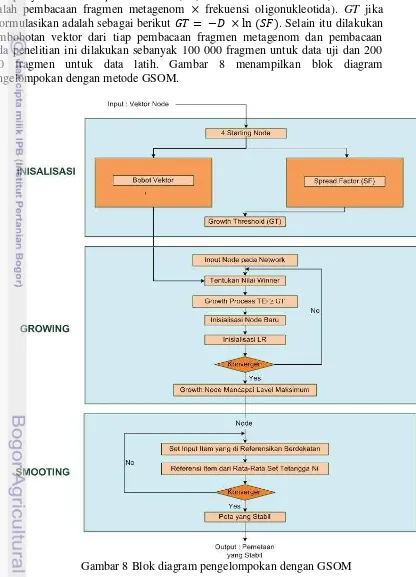
Pengelompokan fragmen metagenom dilakukan dengan GSOM. Arsitektur metode GSOM terdiri dari beberapa fase, yaitu fase inisialisasi, fase growing, dan fase smoothing. Untuk melakukan pengelompokan data, awalnya dilakukan inisialisasi bobot vektor (biasanya di inisialisiasi empat node), nilai Growth Threshold (GT) yang digunakan sebagai batasan dari topologi peta berdasarkan nilai penyebaran atau Spread Factor (SF) dan dimensi dataset D (dimensi D adalah pembacaan fragmen metagenom frekuensi oligonukleotida). GT jika diformulasikan adalah sebagai berikut . Selain itu dilakukan pembobotan vektor dari tiap pembacaan fragmen metagenom dan pembacaan pada penelitian ini dilakukan sebanyak 100 000 fragmen untuk data uji dan 200 000 fragmen untuk data latih. Gambar 8 menampilkan blok diagram pengelompokan dengan metode GSOM.
13
Fase Inisialisasi
Fase pertama dari metode GSOM adalah fase inisialisasi. Empat neuron pertama akan diinisialisasi dengan angka random atau acak. Neuron tersebut diinisialisiasi dengan angka antara 0 sampai 1. Selanjutnya akan dihitung nilai Growth Threshold (GT). Fase inisialisasi digambarkan pada Gambar 9.
Gambar 9 Fase inisialisasi
Perhitungan GT digunakan untuk menentukan dan mendapatkan hasil topologi peta yang ideal. Untuk mendapatkan hasil peta yang ideal, maka harus ditentukan penyebaran dari titik-titik neuron. Pengontrolan ini ditentukan oleh nilai Spread Factor (SF). Nilai SF pada penelitian ini digunakan berbeda pada tiap frekuensi, yaitu 0.6 untuk frekuensi trinukleotida dan spaced k-mer,dan 0.8 untuk frekuensi tetranukleotida
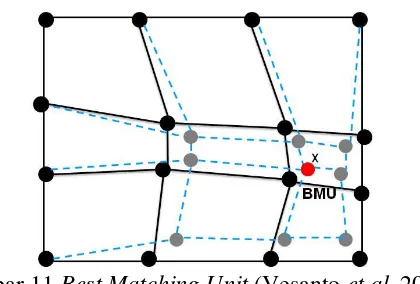
Gambar 10 Inisialisasi starting node
14
While (node hasil inisialisasi bobot) For (tiap epoch)
Tentukan Learning Rate dan Neighborhood Size
If (pemenang yang ditentukan dari matriks komposisi) Then
Bobot vektor diambil dan diaplikasikan pada tetangga dan pemenang itu sendiri
Nilai error dari pemenang ditingkatkan End if
If (total error node i < GT) Grow node jika memenuhi syarat Else
Bobot dari vektor akan didistribusikan pada tetangga sekitar
End if
Inisialisasi Learning Rate dan Neighborhood Size baru Repeat until (semua vektor matriks komposisi digunakan dan grow node mencapai level minimum)
End End
awal ini, semua vektor matriks komposisi akan dipetakan dan mengalami proses growing pada fase selanjutnya, yaitu fase growing.
Fase Growing
Fase growing merupakan fase terpenting dalam metode GSOM karena pada tahap ini peta akan mengalami ekspansi sehingga menjadi lebih dinamik dibandingkan metode SOM. Berikut adalah algoritme fase growing.
Pada fase growing ini juga diinisialisasi beberapa training paramater, yaitu epoch, Learning Rate, dan Neighborhood Size.
Fase Smoothing
Fase smoothing adalah ketika parameter Learning Rate akan di turunkan nilainya begitu juga dengan parameter Neighborhood Size. Learning Rate dan Neighborhood Size yang digunakan akan selalu berubah pada setiap iterasi. Ketika mencapai level yang minimum, maka kedua parameter tersebut juga akan mendekati nilai 0. Untuk penelitian ini, parameter Learning Rate akan di set untuk berhenti pada nilai 0.01 dan Neighborhood Size di set berhenti secara random.
15
Gambar 11 Best Matching Unit (Vesanto et al. 2000)
Titik hitam adalah neuron mula-mula sedangkan adalah vektor yang diambil secara acak. Setelah dilakukan perhitungan jarak maka terlihat perubahan letak neuron yang bergerakn menuju vektor . Pergerakan neuron diilustrasikan dengan titik abu-abu. Titik yang berwarna merah diasumsikan sebagai neuron yang memiliki jarak yang terdekat sehingga disebut dengan BMU.
Evaluasi
Pada evaluasi model untuk pemetaan menggunakan metode GSOM, digunakan dua pengukuran utama, yaitu quantization error dan topographic error. Parameter yang digunakan untuk mendapatkan pengukuran berdasarkan pada parameter learning, topologi peta, dan bentuk dari peta.
Quantization error atau qe (Uriarte dan Martin 2005) adalah pengukuran yang umum digunakan pada penentuan kualitas pemetaan dengan Kohonen map. Pengukuran ini adalah untuk mengukur jarak rata-rata antara vektor data dan Best Matching Unit (BMU) yang berada di sekitar ruang input dan selanjutnya akan mengevaluasi kecocokan dari peta neural, karena itu rata-rata nilai quantization error yang terkecil menunjukkan bahwa jarak vektor data tersebut dekat dengan prototype yang dihasilkan. Perhitungan quantization error, jika diformulasikan adalah sebagai berikut :
Dengan adalah banyak data vektor,
adalah Best Matching prototype
dari vektor data
yang saling berhubungan. Gambar 12 adalah penggambaran pengukuran quantization error.
Gambar 12 Pengukuran quantization error
16
Topographic error atau te (Uriarte dan Martin 2005) digunakan untuk mengukur distorsi error pada topologi peta menggunakan input sampel untuk menentukan pemetaan lanjutan dari ruang input pada grid peta. Jadi BMU yang pertama dan BMU yang kedua bukan merupakan vektor yang saling berdekatan. Topographic error dapat diformulasikan sebagai berikut :
Jika fungsi
adalah 1, maka vektor data
mempunyai BMU yang saling berdekatan. Jika 0, maka bernilai sebaliknya. Diharapkan dengan pengukuran dengan topographic error mendekati nilai 0, yang berarti rata-rata BMU pertama dan BMU kedua tidak saling berdekatan. Gambar 13 menunjukkan pengukuran dengan topographic error.
Gambar 13 Pengukuran topographic error
Selain menggunakan quantization error dan topographic error, digunakan juga persentase error untuk menghitung kesalahan pemetaan pada tiap kelompok. Jadi hanya didasarkan pada perhitungan data fragmen metagenom yang salah pada tiap kelompok filum. Sedangkan untuk uji efisiensi digunakan variabel akurasi kecepatan dalam pengelompokan fragmen metagenom.
Peralatan Penelitian
Alat yang digunakan dalam penelitian ini dibagi dalam perangkat keras dan perangkat lunak, sebagai berikut :
a. Perangkat keras :
Processor : Intel(R) Celeron(R) CPU B815 @ 1.60 GHz 1.60 GHz
Memory : DDR 2 RAM 2 GB
Harddisk : 500 GB b. Perangkat lunak :
Sistem operasi Windows 7 Enterprise 64-bit operating system
Dev CppPortable
Notepad ++
Matlab 7.11.0 (R2010b)
MetaSim version 0.9.1
17
Data simulasi
4 HASIL DAN PEMBAHASAN
Pengelompokan fragmen metagenom dikembangkan dengan bahasa pemrograman C++ dan Matlab 7.11.0 (R2010b). Fragmen metagenom akan dikelompokan dalam 20 kategori, yaitu 20 filum berdasarkan NCBI Taxonomy Browser.
Basis Data Fragmen Metagenom
Penelitian ini menggunakan data fragmen metagenom dengan format FASTA file (FNA) yang diunduh dari situs NCBI dengan memilih 300 mikrob yang nantinya terbagi 200 mikrob untuk data latih dan 100 mikrob untuk data uji. Data mikrob untuk data latih dan data uji yang digunakan pada penelitian ini dapat dilihat pada Lampiran 1 dan Lampiran 2. Data latih digunakan untuk mendapatkan model pengelompokan berdasarkan tingkat filum. Sedangkan data uji digunakan untuk mengevaluasi model pengelompokan. Selain itu data uji akan dilakukan pengujian pada parameter yang berbeda untuk mendapatkan ukuran peta yang terbaik berdasarkan hasil evaluasi error terkecil menggunakan tiga frekuensi oligonukleotda.
Ekstraksi Ciri dengan K-Mer Frequency
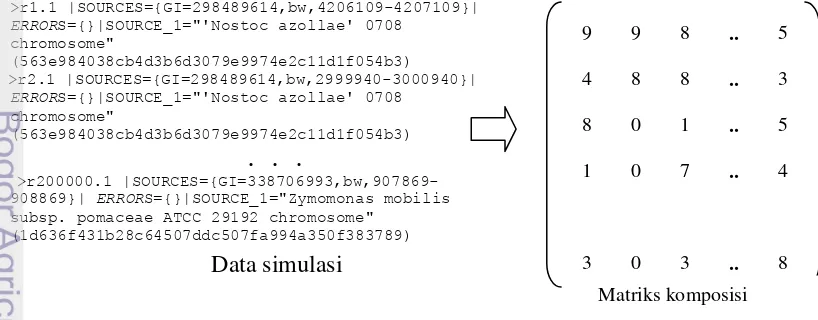
Fragmen metagenom hasil simulasi MetaSim akan diekstraksi dengan k-mer frequency. Ekstraksi dengan k-mer akan membentuk matriks komposisi sesuai dengan berapa banyak data yang dibangkitkan dan frekuensi oligonukleotida yang digunakan. Frekuensi fragmen metagenom yang diekstraksi dengan k-mer frequency adalah trinukleotida dan tetranukleotida. Banyak data yang dibangkitkan adalah 200 000 untuk data latih dan 100 000 untuk data uji. Fitur yang digunakan adalah sebanyak 64 untuk trinukleotida, dan 256 untuk tetranukleotida. Sehingga didapat perhitungan untuk tiap frekuensi oligonukleotida akan diperoleh matriks komposisi dengan ukuran , , , dan ,; masing-masing untuk data latih dan data uji. Contoh hasil ekstraksi ciri pada salah satu frekuensi oligonukleotida ditunjukkan pada Gambar 14.
>r1.1 |SOURCES={GI=298489614,bw,4206109-4207109}|
908869}| ERRORS={}|SOURCE_1="Zymomonas mobilis
subsp. pomaceae ATCC 29192 chromosome" (1d636f431b28c64507ddc507fa994a350f383789)
Gambar 14 Matriks komposisi salah satu frekuensi oligonukleotida
18
Ekstraksi Ciri dengan Spaced k-mer
Selain menggunakan k-mer frequency untuk ekstraksi ciri, digunakan spaced k-mer. Ekstraksi dengan spaced k-mer lebih ekonomis dilihat dari sisi penerimaan informasi (information retrieval) karena ekstraksi ini menggunakan kondisi don’t care sehingga waktu yang dibutuhkan tidak terlalu lama tapi sudah mendapatkan informasi tentang komposisi dari fragmen metagenom dengan lebih terperinci. Data fragmen metagenom dihitung hampir sama dengan menggunakan k-mer frequency, tapi ekstraksi ini memperhatikan don’t care yang mempunyai pola , dengan adalah kondisi don’t care. Sehingga dari perhitungan didapat dimensi fitur adalah sebanyak 192. Ukuran matriks komposisi dengan ekstraksi spaced k-mer pada data latih adalah dan 1 untuk data uji.
Praproses Data Fragmen Metagenom
Praproses bertujuan untuk mengurangi variasi data sehingga data mudah untuk dipahami. Fragmen metagenom yang awalnya berupa matriks komposisi diubah menjadi data matriks yang bernilai antara 0 dan 1 dengan metode decimal scaling. Contoh hasil decimal scaling yang diperoleh untuk salah satu frekuensi oligonukleotida ditunjukkan pada Gambar 15.
9 9 8 .. 5
Data Simulasi Hasil Decimal Scaling
Gambar 15 Matriks decimal scaling salah satu frekuensi oligonukleotida
Pembagian Data Latih dan Data Uji
19 Tabel 3 Pembagian mikrob data latih dan data uji
Data latih Data uji
No Mikrob No Mikrob
1 Acetobacterium woodi DSM 1030
chromosome
1 Acaryochloris marina MBIC11017 chromosome
2 Acidaminococcus fermentans DSM
20731 chromosome
2 Acetobacter pasterianus IFO 3283-01
3 Acidithiobacillus ferrivorans SS3 chromosome
3 Acholeplasma laidlawii PG-8A
chromosome
4 Acidovorax sp.JS42 chromosome 4 Acidimicrobium ferroxidans DSM
10331 chromosome
5 Acinetobacter sp.ADP1 chromosome 5 Actinobacillus pleuropneumoniae
serovar 3 str. JL03 chromosome
. . . . . .
200 Zymomonas mobilis subsp.pomaceae ATCC 29192 chromosome
100 Weissella korensis KACC 15510 chromosome
Data fragmen metagenom, masing-masing data latih dan data uji akan di bangkitkan sebanyak 200 000 fragmen untuk data latih dan 100 000 fragmen untuk data uji. Banyaknya pembangkitan data dari tiap kelompok filum dihitung secara otomatis ketika data disimulasi oleh MetaSim untuk setiap mikrob. Hasil perhitungan pembangkitan data latih dan data uji ditampilkan pada Tabel 4 dan Tabel 5.
Tabel 4 Pembangkitan data latih
No Filum Reads
6 Verrucomicrobia 4679
7 Chloroflexi 13 760
8 Cyanobacteria 16 376
9 Deinococcus-thermus 7606
10 Acidobacteria 10 781
11 Firmicutes 17 559
12 Fusobacteria 3400
13 Gemmatimonadetes 1484
14 Nitrospirae 2831
15 Planctomycetes 10 830
16 Proteobacteria 18 984
17 Spirochaetes 8702
18 Synergistetes 1922
19 Tenericutes 11 651
20
Tabel 5 Pembangkitan data uji
No Filum Reads
1 Actinobacteria 5452
2 Aquificae 2144
3 Bacteroidetes 5330
4 Chlorobi 3950
5 Chlamydiae 3764
6 Verrucomicrobia 3716
7 Chloroflexi 8652
8 Cyanobacteria 5685
9 Deinococcus-thermus 3873
10 Acidobacteria 10 199
11 Firmicutes 7648
12 Fusobacteria 3281
13 Gemmatimonadetes 1398
14 Nitrospirae 2751
15 Planctomycetes 9168
16 Proteobacteria 12 518
17 Spirochaetes 5829
18 Synergistetes 1846
19 Tenericutes 999
20 Thermotogae 1797
Pengelompokan Fragmen Metagenom dengan GSOM
Frekuensi oligonukleotida adalah frekuensi kemunculan pasangan basa pada fragmen metagenom, dan pada penelitian ini muncul sebanyak trinukleotida, tetranukleotida, dan menggunakan frekuensi spaced k-mer yang memperhatikan kondisi don’t care. Dalam penelitian ini, kemunculan frekuensi trinukleotida pada fragmen metagenom adalah sebanyak 64 fitur, frekuensi tetranukleotida sebanyak 256 fitur, dan frekuensi spaced k-mer sebanyak 192 fitur.
Fase Inisialisasi
Fase inisialisasi merupakan fase awal untuk menentukan parameter global, yaitu GrowthThreshold (GT). Parameter ini ditentukan oleh pengguna (user). GT digunakan untuk mengatur penyebaran neuron pada peta. Untuk frekuensi trinukleotida, parameter GT diset dengan nilai 0.6. Untuk tetranukleotida diset dengan nilai 0.8, dan 0.6 untuk frekuensi spaced k-mer. Insialisasi bobot menggunakan inisialisasi secara random atau acak. Setiap data vektor akan diberi nilai hasil distribusi antara nilai yang paling minimum dan yang paling maksimal dari dataset fragmen metagenom.
Fase Growing
21
Matriks hasil inisialisasi bobot pada trinukleotida
0.0347 0.0551 0.0929 . . . 0.2122
Matriks hasil inisialisasi bobot pada tetranukleotida
0.0801 0.1864 0.1055 . . . 0.3639
Matriks hasil inisialisasi bobot pada frekuensi spaced k-mer
Gambar 16 Insialisasi bobot pada frekuensi oligonukleotida
Fase Smoothing
Pada fase ini, semua hasil pelatihan dan pengujian akan berhenti mengalami proses growing. Hasil pengelompokan akan menghasilkan pemetaan yang stabil dan tiap data vektor akan dipetakan pada grid peta. Gambar 17, Gambar 18 dan Gambar 19 menampilkan hasil pemetaan frekuensi trinukleotida, tetranukleotida dan spaced k-mer berdasarkan algoritme GSOM.
Evaluasi
22
Gambar 17 Pemetaan frekuensi trinukleotida
23
Gambar 19 Pemetaan frekuensi spaced k-mer Frekuensi Trinukleotida
24
Tabel 6 Perhitungan quantization error pada trinukleotida
LR NS
0 1 2 3 4
0,1 0,759 0,531 0,709 0,76 0,758
0,25 0,744 0,752 0,741 0,742 0,747
0,5 0,639 0,736 0,639 0,742 0,741
0,75 0,871 0,757 0,667 0,752 0,684
0,9 0,842 0,834 0,781 0,773 0,794
Tabel 7 Perhitungan topographic error pada trinukleotida
LR NS
0 1 2 3 4
0,1 0,125 0,101 0,119 0,124 0,134
0,25 0,112 0,127 0,129 0,117 0,132
0,5 0,109 0,115 0,117 0,11 0,134
0,75 0,108 0,109 0,131 0,111 0,135
0,9 0,109 0,109 0,132 0,124 0,134
Tabel 8 Perhitungan persentase error pada trinukleotida
LR NS
0 1 2 3 4
0,1 18,73 16,84 19,21 18,83 19,03
0,25 17,69 18,63 17,09 18,23 18,76
0,5 18,2 18,08 16,97 18,37 18,41
0,75 18,02 18,88 17,79 18,74 18,34
0,9 18,44 18,34 19,02 19,23 18,24
Dari Tabel 6, Tabel 7 dan Tabel 8, kombinasi parameter yang memberikan hasil error yang paling kecil adalah pada Learning Rate 0.1 dengan Neighborhood Size 1. Hasil error terkecil yang diberikan adalah 0.531 untuk quantization error dan 0.101 untuk topographic error. Sedangkan untuk kesalahan pengelompokan memberikan hasil error sebesar 16.84%.
25
Gambar 20 Perhitungan quantization error pada trinukleotida
Gambar 21 Perhitungan topographic error pada trinukleotida
0
Perhitungan Quantization Error pada trinukleotida
LR 0.1
Perhitungan Topographic Error pada trinukleotida
26
Gambar 22 Perhitungan persentase error pada trinukleotida
Frekuensi Tetranukleotida
Hasil kombinasi atau variasi parameter pada frekuensi tetranukleotida dilakukan berdasarkan perhitungan error, yaitu quantization error, topographic error, dan persentase error. Tabel 9, Tabel 10 dan Tabel 11 menampilkan error yang dihasilkan untuk tiap kombinasi parameter Learning Rate (LR) dan Neighborhood Size (NS). Kolom yang berwarna abu-abu menunjukkan nilai error terkecil pada frekuensi tetranukleotida.
Tabel 9 Perhitungan quantization error pada tetranukleotida
LR NS
Tabel 10 Perhitungan topographic error pada tetranukleotida
LR NS
Perhitungan Persentase Error pada trinukleotida
27 Tabel 11 Perhitungan persentase error pada tetranukleotida
LR NS memberikan hasil error yang paling kecil adalah pada Learning Rate 0.75 dengan Neighborhood Size 1. Hasil error terkecil yang diberikan adalah 0.886 untuk quantization error dan 0.09 untuk topographic error. Sedangkan untuk kesalahan pengelompokan memberikan hasil error sebesar 15.43%.
Hasil kombinasi paramater pada frekuensi tetranukleotida dapat dilihat pada Gambar 23 untuk perhitungan quantization error, dan Gambar 24 untuk perhitungan topographic error. Untuk kesalahan dalam pengelompokan, ditunjukkan pada Gambar 25.
Gambar 23 Perhitungan quantization error pada tetranukleotida
0,000
Perhitungan Quantization Error pada tetranukleotida
28
Gambar 24 Perhitungan topographic error pada tetranukleotida
Gambar 25 Perhitungan persentase error pada tetranukleotida
0
Perhitungan Topographic Error pada tetranukleotida
LR 0.1
Perhitungan Persentase Error pada tetranukleotida
29
Frekuensi Spaced k-mer
Hasil kombinasi atau variasi parameter pada frekuensi spaced k-mer dilakukan berdasarkan perhitungan error, yaitu quantization error, topographic error, dan persentase error. Tabel 12, Tabel 13 dan Tabel 14 menampilkan error yang dihasilkan untuk tiap kombinasi parameter Learning Rate (LR) dan Neighborhood Size (NS). ). Kolom yang berwarna abu-abu menunjukkan nilai error terkecil pada frekuensi spaced k-mer.
Tabel 12 Perhitungan quantization error pada spaced k-mer
LR NS
Tabel 13 Perhitungan topographic error pada spaced k-mer
LR NS
Tabel 14 Perhitungan persentase error pada spaced k-mer
LR NS memberikan hasil error yang paling kecil adalah pada Learning Rate 0.5 dengan Neighborhood Size 1. Hasil error terkecil yang diberikan adalah 0.665 untuk quantization error dan 0.06 untuk topographic error. Sedangkan untuk kesalahan pengelompokan memberikan hasil error sebesar 13.07%.
30
Gambar 26 Perhitungan quantization error pada spaced k-mer
Gambar 27 Perhitungan topographic error pada spaced k-mer 0,000
Perhitungan Quantization Error pada Spaced K-Mer
LR 0.1
Perhitungan Topographic Error pada Spaced K-Mer
31
Gambar 28 Perhitungan persentase error pada spaced k-mer
Berdasarkan hasil pertimbangan error yang dihasilkan dari ketiga frekuensi oligonukleotida, maka frekuensi spaced k-mer memberikan hasil error yang paling kecil dari perhitungan kombinasi parameter. Untuk quantization error, frekuensi spaced k-mer memberikan hasil 0.665 dan hasil error tersebut masih lebih besar dibandingkan dengan frekuensi trinukleotida yang hanya memberikan hasil quantization error 0.531.
Meskipun demikian, jika dilihat dari distorsi error pada topologi peta menggunakan pengukuran topographic error, frekuensi spaced k-mer memberikan error yang lebih kecil dibandingkan dengan trinukleotida dan tetranukleotida, yaitu 0.06 sehingga menjadikan frekuensi spaced k-mer lebih baik dalam pemetaan fragmen metagenom dibandingkan dengan frekuensi oligonukleotida yang lain.
Selain dilihat dari hasil pengukuran quantization error dan topographic error, persentase error dalam mengelompokan fragmen metagenom menggunakan frekuensi spaced k-mer menunjukkan hasil error yang paling rendah, yaitu 13.07%. Hal ini menunjukkan bahwa kombinasi parameter Learning Rate 0.5 dengan Neighborhood Size 1 pada frekuensi spaced k-mer memberikan hasil terbaik dalam pengelompokan fragmen metagenom dengan metode GSOM.
Dari kombinasi paramater terbaik pada frekuensi spaced k-mer, maka akan dilihat pengujian pada map size dari [100 – 500] dengan mengkombinasikan unit dari peta untuk mendapatkan kombinasi map size dan unit peta terbaik dalam pemetaan fragmen metagenom dengan metode GSOM. Tabel 15 akan menampilkan parameter yang digunakan dan Tabel 16 menunjukkan hasil pelatihan dari parameter yang digunakan.
12
Perhitungan Persentase Error pada Spaced K-Mer
32
Training length 10 epochs
Tabel 16 Hasil pelatihan frekuensi spaced k-mer
Map size Unit peta Waktu latih Persentase error
[100 100] 300 42 menit 6.73%
Tabel 16 menunjukkan hasil dari pelatihan fragmen mentagenom dengan metode GSOM pada frekuensi spaced k-mer. Pada data yang memiliki map size [100 100] dan [100 150] dengan unit peta sebanyak 300 unit memiliki persentase error yang kecil, yaitu 6.73% dan 6.43%. Sedangkan untuk data yang memiliki map size yang besar seperti [500 500] dengan unit peta sebanyak 5000 unit, memberikan hasil persentase error yang lebih banyak, yaitu 10.14%.
Hasil pengujian tersebut menunjukkan bahwa map size dan unit dari peta berpengaruh pada waktu pelatihan dan juga persentase error yang dihasilkan. Semakin kecil map size dan unit peta, maka makin sedikit waktu yang dibutuhkan untuk pelatihan dan persentase error yang dihasilkan. Dari hasil tersebut maka dapat diketahui bahwa metode GSOM memiliki akurasi dalam pengelompokan yang baik dengan menggunakan frekuensi spaced k-mer dengan persentase error ≥ 89.71% dengan menggunakan map size [100 – 500] dan unit peta dari 300 – 5000 unit peta.
Pengujian Data Menggunakan BLAST
Pada penelitian ini dilakukan pengujian pada mikrob yang tidak termasuk pada data uji, yaitu mikrob Bacteroides fragilis 638R dengan menggunakan panjang fragmen 1 kbp. Banyak pembacaan adalah 1024 reads. Kelompok filum mikrob tersebut adalah Bacteroidetes. Hasil pengelompokan sebagian besar mengelompokan pada filum Bacteroidetes. Hasil pengelompokan dapat dilihat pada Gambar 29.
33 dapat dilihat bahwa kelompok filum Bacteroidetes memiliki kesamaan yang tinggi dengan Bacteroides fragilis 638R.
Gambar 29 Hasil pengelompokan Bacteroides fragilis 638R dengan 1024 reads Berikut adalah hasil dari alignment BLAST untuk mencari kesamaan mikrob Bacteroides fragilis 638R. Gambar 30 memperlihatkan data yang digunakan, Gambar 31 menunjukkan ‘hit’ dari pencarian BLAST. Tabel 17 menampilkan 17 mikrob yang memiliki kesamaan dengan Bacteroides fragilis 638R pada pencarian kesamaan menggunakan BLAST. Lampiran 6 menunjukkan pohon taksonomi BLAST dari organisme yang memiliki kesamaan dengan Bacteroides fragilis 638R.
Gambar 30 Data yang digunakan dengan panjang sekuens query 1000 (1 kbp)
94,24%
4,49% 1,27%
Bacteroidetes
Chlamydiae
Proteobacteria
Bacteroidetes
34
Gambar 31 Hit dari 17 organisme yang memiliki kesamaan dengan Bacteroides fragilis 638R
Tabel 17 Daftar organisme yang memiliki kesamaan dari hasil alignment Bacteroides fragilis 638R pada BLAST
No Deskripsi Max 1 Bacteroides fragilis 638R
genome
1804 1804 100% 0.0 100%
2 Bacteroidesfragilis NCTC 9343, complete genome
1804 1804 100% 0.0 100%
3 Bacteroides fragilis YCH46 DNA, complete genome
1790 1790 100% 0.0 99%
4 Uncultured organism clone 1041059767817genomic sequence
250 250 83% 2e-62 67%
5 Uncultured orgnism clone VC1AB77TF genomic sequence
199 199 47% 4e-47 70%
6 Bacteroides helcogenes P 36-108, complete genome
168 168 85% 6e-38 66%
7 Tannerella forsythia ATCC 43037, complete genome
9 Melioribacter roseus P3M, complete genome
35
No Deskripsi Max
score Total score
Query cover
E value
Max ident 14 Bacteroides vulgatus ATCC
8482, complete genome
46.4 46.4 13% 0.52 69%
15 Prevotella denticola F0289, complete genome
42.8 42.8 9% 6.3 70%
16 Prevotella melaninogenica ATCC 25845 chromosome I, complete sequence
42.8 42.8 9% 6.3 70%
17 Bacteroides
thetaiotaomicron VPI-5482, complete genome
42.8 42.8 6% 6.3 75%
36
5 SIMPULAN DAN SARAN
Simpulan
Simpulan dari hasil penelitian ini adalah :
1. Metode GSOM dapat digunakan untuk pemetaan fragmen metagenom yang memiliki komunitas yang besar dan memiliki panjang fragmen yang pendek, yaitu 1 kbp.
2. Pengelompokan fragmen metagenom dengan metode GSOM menghasilkan
akurasi ≥ 80%, sehingga dapat dikatakan bahwa metode GSOM dapat mengelompokan fragmen metagenom dengan benar dengan kombinasi parameter terbaik adalah menggunakan Learning Rate sebesar 0.5 dengan Neighborhood Size sebesar 1.
3. Untuk frekuensi oligonukleotida, frekuensi spaced k-mer merupakan frekuensi yang terbaik untuk memetakan fragmen metagenom karena menghasilkan persentase error hanya sebesar 13.07% dengan distorsi error sebesar 0.06 untuk pemetaan dengan map size [10 10].
4. Pelatihan dengan menggunakan frekuensi spaced k-mer dengan kombinasi parameter terbaik menghasilkan pemetaan terbaik pada map size [100 150] dengan unit peta sebanyak 100 unit. Persentase error yang dihasilkan adalah 6.43% dengan waktu latih 51 menit.
5. Banyaknya unit peta yang digunakan dan map size mempengaruhi waktu pelatihan dan persentase error yang dihasilkan. Semakin sedikit unit peta yang digunakan dan makin kecil map size, maka waktu pelatihan yang dibutuhkan makin cepat dan persentase error yang dihasilkan makin kecil.
Saran
Adapun saran untuk penelitian selanjutnya adalah :
1. Menggunakan data yang riil sehingga tidak lagi menggunakan data hasil simulasi
37
DAFTAR PUSTAKA
Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuku T, Ikemura T. 2003. Informatics for unveiling hidden genome signatures. Genome Research. 179(4):693-701. doi:10.1101/gr.634603
Amano K, Nakamura H, Ichikawa H. 2003. Self-organizing clustering : a novel non-hierarchical method for clustering large amountof sequece DNAs. Genome Informatics. 14: 575-576
Amano K, Nakamura H, Ichikawa H, Numa H, Kobayashi KF, Nagamura Y, Onodera N. 2007. Self-organizing clustering : non-hierarchical clustering for large-scale sequence DNA data. IPSJ Digital Courier. 2(2):523-527 Brady A, Salzberg SL. 2009.Phymm and phymmbl : metagenomic phylogenetic
classification with interpolated markov models. Nature Methods. 6 (9) : 673
– 676. doi : 10.1038/nmeth.1358
Chan CK, Hsu AL, Tang SL, Halgamuge SK. 2007. Using growing self-organizing maps to prove the binning process in environmental whole-genome shotgun equencing. Journal of Biomedicine and Biotechnology. 2008. doi:10.1155/2008/513701
Choi JH, Cho HG. 2002. Analysis of common k-mers for whole genome sequence using SSB-tree. Genome Information. 13 : 30-41
De Silva D, Alahakoon D, Dharmage S. 2007. Cluster analysis using the GSOM : patterns in epidemiology. IEEE International Conference on ICIAF. 5(7):63
– 69. doi : 10.1109/ICIAFS.2007.4544781
Federhen S. 2012. The NCBI taxonomy database. Nucleic Acids Research. 40: 136- 143. doi : 10.1093/nar/gkr1178
Han J, Kamber M, Pei J. 2012. Data mining concepts and techniques. 3th ed. Waltham (US) : Morgan Kaufmann Publishers
Harayama S, Kasai Y, Hara A. 2004. Microbial communities in oil-contaminated seawater. Current Opinion in Biotechnology. 15:205-214
Hsu AL, Halgamuge SK. 2002. Enhancement of topology preservation and hierarchical dynamic self-organizing maps for data visualisation. International Journal of Approximate Reasoning. 32(2003):259-279
Hsu Al, Tang SL, Halgamuge SK. 2003. An unsupervised hierarchical dynamic self-organizing approach to cancer class discovery and marker gene identification in microarray data. Bioinformatics. 19(16) : 2131-2140. doi : 10.1093/bioinformatics.btg296
Huson DH, Auch AF. Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Research. 17 : 1 – 11. doi : 10.1101/gr/5969107
Kusuma WA. 2012. Combined approaches for improving the performance of de novo dna sequence assembly and metagenomic classification of short fragments from next generation sequencer [disertasi]. Tokyo (JP) : Tokyo Institute of Technology.
Meyerdierks A, Glockner FO. 2010. Metagenome analysis. Advances in Marine Genomics. 1 : 33 – 71. doi : 10.1007/978-90-481-8639-6_2
38
Nasser S, Brelan A, Harris FC, Nicolescu M. 2008. A fuzzy classifier to taxonomically group dna fragments within a metagenome. Annual Meeting of the NAFIPS 08. 8 : 1-6
O’Malley M. 2012. Metagenomics. Springer [Internet].[diunduh 2012 Nov 29]. Tersedia pada : http://www.maureenomalley.org/publications.html
Pati A, Heath LS, Kyrpides NC, Ivanova N. 2011. ClaMS : A classifier for metagenomic sequences. Standards in Genomic Science. 5 : 248 – 253. doi :10.4056/sigs.2075298
Prabhakara S, Acharya R. 2012. Unsupervised two-way clustering of metagenomic sequence. Journal of Biomedicine and Biotechnology. doi : 101.1155/2012/153647
Richter DC, Ott F, Auch AF, Schmid R, Hudson DH. 2008. MetaSim-sequencing simulator for genomics and metagenomics. PLoS ONE. 3(10). doi:10.1371/journal.pone.0003373
Rodriguez AA, Bompada T, Syed M, Shah PK, Maltsev N. 2007. Evolutionary analysis of enzymes using chisel. Bioinformatics. 23( 22)
Rosen G, Garbarine E, Caseiro D, Polikar R, Sokhansanj B. 2008. Metagenome fragment classification using n-mer frequency profiles. Advances in Boinformatics. doi : 10.1155/2008/205969
Sheaffer RL, Mendenhall W, Ott RL. 1990. Elementary survey sampling. 4th ed. Boston (US) : PWS – KENT Publishing Company
Teeling H, Waldmann J, Lombardot T, Bauer M, Glockner FO. 2004. TETRA : a web service and stand-alone program for the analysis and comparison of tetranucleotide usage pattern in sequence DNAs. BMC Informatics. 5(163). doi:10.1186/1471-2105-5-163
Uriarte EA, Martin FD. 2005. Topology preservation in SOM. International Journal of Applied Mathematics and Computer Sciences. 1(1) : 19 - 22 Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D,
Paulsen I, Nelson KE, Nelson W et al. 2004. Environmental genome shotgun sequencing of the sargasso sea. Science. 304 : 66 – 74. doi : 10.1126/science.1093857
Vesanto J, Himberg J, Alhoniemi E, Parhankangas J. 2000.SOM toolbox for matlab 5. Helsinski University of Technology. [Internet].[diunduh 2013 Jan 10]. Tersedia pada : http:///www.cis.hut.fi/projects/somtoolbox/
Woyke T, Teeling H, Ivanova NN, Hunteman M, Richter M, Gloeckner FO, Boffelli D, Anderson IJ, Barry KW, Shapiro HJ et al. 2006. Symbiosis insights through metagenomic analysis of a microbial consortium. Nature. 443(7114): 950-5.
Wu H. 2008. PCA – based Linear Combinations of Oligonucleotide Frequencies for Metagenomic DNA Fragment Binning. IEEE Symposium on CIBCB. 8 (2008): 46-53
Wu X, Lee W, Tseng C. 2005. ESTmapper : efficiently aligning sequence DNAs to genomes. IEEE International Paralel and Distributed Processing Symposium. 204(2005) : 196 – 204. Doi : 10.1109/IPDPS.2005.204
40
Lampiran 1Daftar mikrob yang digunakan sebagai data latih
Reads Mikrob
1702 Reads `'Nostoc azollae' 0708 chromosome'
1283 Reads `Acetobacterium woodii DSM 1030 chromosome'
716 Reads `Acidaminococcus fermentans DSM 20731 chromosome'
1053 Reads `Acidithiobacillus ferrivorans SS3 chromosome'
1391 Reads `Acidovorax sp. JS42 chromosome'
1097 Reads `Acinetobacter sp. ADP1 chromosome'
1460 Reads `Advenella kashmirensis WT001 chromosome'
1133 Reads `Aequorivita sublithincola DSM 14238 chromosome'
865 Reads `Akkermansia muciniphila ATCC BAA-835 chromosome'
1186 Reads `Alistipes finegoldii DSM 17242 chromosome'
661 Reads `Aminobacterium colombiense DSM 12261 chromosome'
658 Reads `Anaerobaculum mobile DSM 13181 chromosome'
1118 Reads `Anaerolinea thermophila UNI-1'
525 Reads `Aquifex aeolicus VF5'
650 Reads `Arcanobacterium haemolyticum DSM 20595 chromosome'
1318 Reads `Belliella baltica DSM 15883 chromosome'
195 Reads `Blattabacterium sp. (Blaberus giganteus) chromosome'
199 Reads `Blattabacterium sp. (Blattella germanica) str. Bge'
199 Reads `Blattabacterium sp. (Cryptocercus punctulatus) str. Cpu chromosome'
184 Reads `Blattabacterium sp. (Mastotermes darwiniensis) str. MADAR chromosome'
218 Reads `Blattabacterium sp. (Periplaneta americana) str. BPLAN'
146 Reads `Buchnera aphidicola (Cinara tujafilina) chromosome'
188 Reads `Buchnera aphidicola str. Bp (Baizongia pistaciae) chromosome'
154 Reads `Buchnera aphidicola str. Cc (Cinara cedri)'
1176 Reads `Burkholderia ambifaria AMMD chromosome 1'
1055 Reads `Burkholderia cenocepacia HI2424 chromosome 1'
1268 Reads `Burkholderia glumae BGR1 chromosome 1'
1641 Reads `Caldilinea aerophila DSM 14535 = NBRC 104270'
370 Reads `Candidatus Azobacteroides pseudotrichonymphae genomovar. CFP2 chromosome'
884 Reads `Candidatus Chloracidobacterium thermophilum B chromosome chromosome 1'
1390 Reads `Candidatus Nitrospira defluvii'
764 Reads `Candidatus Protochlamydia amoebophila UWE25 chromosome'
3274 Reads `Candidatus Solibacter usitatus Ellin6076 chromosome'
799 Reads `Capnocytophaga canimorsus Cc5 chromosome'
853 Reads `Capnocytophaga ochracea DSM 7271 chromosome'
3373 Reads `Catenulispora acidiphila DSM 44928 chromosome'
1567 Reads `Cellulophaga algicola DSM 14237 chromosome'
385 Reads `Chlamydia muridarum Nigg'
351 Reads `Chlamydia trachomatis 434/Bu chromosome'
329 Reads `Chlamydia trachomatis A/HAR-13'
41
Reads Mikrob
342 Reads `Chlamydia trachomatis B/Jali20/OT chromosome'
342 Reads `Chlamydia trachomatis B/TZ1A828/OT chromosome'
341 Reads `Chlamydia trachomatis D-EC chromosome'
340 Reads `Chlamydia trachomatis L2b/UCH-1/proctitis chromosome'
351 Reads `Chlamydophila abortus S26/3'
1017 Reads `Chlorobium phaeobacteroides DSM 266 chromosome'
642 Reads `Chlorobium phaeovibrioides DSM 265 chromosome'
1571 Reads `Chloroflexus aggregans DSM 9485 chromosome'
1717 Reads `Chloroflexus sp. Y-400-fl chromosome'
1179 Reads `Coraliomargarita akajimensis DSM 45221 chromosome'
867 Reads `Corynebacterium aurimucosum ATCC 700975'
816 Reads `Corynebacterium diphtheriae 241 chromosome'
1010 Reads `Corynebacterium efficiens YS-314 chromosome'
1095 Reads `Corynebacterium glutamicum ATCC 13032'
827 Reads `Corynebacterium jeikeium K411 chromosome'
809 Reads `Corynebacterium kroppenstedtii DSM 44385 chromosome'
770 Reads `Corynebacterium pseudotuberculosis 267 chromosome'
2019 Reads `Cyclobacterium marinum DSM 745 chromosome'
460 Reads `Dehalococcoides ethenogenes 195'
532 Reads `Dehalogenimonas lykanthroporepellens BL-DC-9 chromosome'
809 Reads `Deinococcus geothermalis DSM 11300'
2278 Reads `Dyadobacter fermentans DSM 18053 chromosome'
601 Reads `Fervidobacterium nodosum Rt17-B1 chromosome'
1139 Reads `Flavobacterium branchiophilum FL-15'
1012 Reads `Flavobacterium columnare ATCC 49512 chromosome'
946 Reads `Flavobacterium indicum GPTSA100-9'