Kajian Molekular Mutant Del
-SEAThalassemia-
α
pada Populasi Medan
Ratna Akbari Ganie
Departemen Patologi Klini, Fakultas Kedokteran, Universitas Sumatera Utara, Medan
Abstrak: Jumlah total dari 1.521 merupakan individu dewasa sehat dari tujuh kelompok etnik
berbeda yang hidup di kota Medan yang tersaring pembawa α thalessemia dengan hemoglobin
electrophoresis. Diantara semua sampel, 51 orang dicurigai terdapat ciri thalassemia, mutasi DNA
dideteksi dengan metode PCR. Mutasi 21 del-SEA
ditemukan pada suku Aceh sebanyak 3.5%, 2.97% di suku Melayu, 2,69% Cina, 1,71% suku Jawa, 1.19% suku Batak dan tidak ditemukan pada suku Minangkabau dan Nias. Mutasi 21 del-SEA
biasanya juga ditemukan pada populasi di Asia Tenggara dan Pasifik atau gen Mongoloid dan Malenesian sepanjang 17.5 kb pada gen globin α1 maupun α1. Penggolongan subjek homozigot dengan cara menurunkan atau menghilangkan gen globin pada hipokromik, anemia hemolitik, dan ketergantungan pada tranfusi darah untuk memperpanjang hidup orang yang terjangkit thalassemia-α. Berdasarkan pada pembiayaan ini, dapat dipahami bahwa semua kelompok etnik yang ada di Medan secara genetik termasuk ke dalam bagian gen Mongolia dan Malanesia.
Kata kunci: α-thalassemia, mutan del-SEA, kelompok etnik di Medan
Abstract: A total number of 1.521 apparently healthy adult individuals from seven mayor different ethnic groups who habited Medan were screened for α-thalassemia carrier by hemoglobin electrophoresis. Among these, 51 subjects were suspected as α-thalassemia trait; the
DNA mutation were determined using PCR method to detect the del-SEA
mutation performance. During this study, 21 del-SEA
mutations have been identified by specific olygonucleotides primers in descending frequencies, 3.5% in Aceh; 2.97% in Malay; 2,69% in Chinese; 1.71% in Javanese;
1,19% in Batak and absent in Minang and Nias. The del-SEA
mutant was commonly found in Southeast Asian and Pacific populations or Mongoloid and Melanesian gene pools result in about 17.5 Kb deletion along the α1 and α2 globin genes. The homozygote subject characterized by
reduced or absent of α-globin gene expression leading to hypochromic, hemolytic anemia and dependence on blood transfusions to sustain life which was known as thalassemia-α. Based on this funding, understandable that these Medan’s ethnic groups genetically share the Mongoloid and Melanesian gene pools.
Keywords: α-thalassemia, del-SEA mutant, Medan’s ethnic group
PENDAHULUAN
Seperti daerah endemik malaria lainnya, keberadaan penyakit thalassemia dan hemoglobinopatia pada penduduk Indonesia cukup tinggi, sebagai seleksi positif terhadap invasi plasmodium (Flint et al., 1993).1
Dari beberapa penelitian terdahulu telah dilaporkan bahwa keberadaan thalassemia-β dan Hemoglobin-E cukup tinggi baik dalam skala sporadis maupun nasional, bahkan pada beberapa populasi frekuensinya mencapai >15%. Sementara itu, penelitian tentang keberadaan thalassemia-α
masih sangat terbatas, walaupun telah dilaporkan oleh Hill et al., (1989) bahwa prevalensi mutasi
yang menyebabkan gangguan sintesis globin-α
ini cukup tinggi pada populasi Asia Daratan dan Pasifik.2
WHO (1994) memperkirakan terdapat kira-kira 13.000–16.000 bayi thalassemia-α baru, lahir setiap tahunnya di seluruh dunia dan jika mereka dapat mencapai usia dewasa maka diperkirakan ada 680.000 penderita thalassemia-α di Asia Tenggara (Higgs, 1983).3,4
Prevalensi thalasemia-α
Sementara itu, angka yang banyak dirujuk para ahli epidemiologi genetic Indonesia adalah estimasi Wong (1983) yang memperkirakan terdapat kira-kira 0,5% carrier thalassemia-α di Indonesia secara nasional, jauh di bawah angka pembawa sifat thalassemia-β yang diperkirakan mencapai 3,5% dan Hb-E yang mencapai 5%.7
Nanum demikian banyak peneliti percaya bahwa angka thalassemia-α jauh di atas angka tersebut, bahkan pada beberapa populasi Indonesia di Jawa, Kalimantan, dan Sulawesi yang telah diteliti mencapai 2,5- 3-2% (Setianingsih, 2003).8
Dugaan tersebut diperkuat dengan keberadaan kasus bayi Hydrop Fetalis dan Hb-H yang dijumpai di rumah sakit-rumah sakit rujukan cukup tinggi. Walaupun keberadaan thalassemia-α di Medan telah pernah dilaporkan sebelumnya oleh Hariman et al.(1984) yaitu masing-masing sebesar 2,5% untuk thalassemia-αo
dan thalassemia-α+
, tetapi masih terbatas pada aras Biokimiawi melalui skrining hematologis.9
Penelitian tingkat molekular untuk mengetahui dasar molekular penyakit tersebut pada populasi Medan sampai saat ini belum pernah dilakukan. Sebelumnya telah dilaporkan terdapat setidaknya 37 jenis mutan pada gen globin-α1 dan globin-α2 sebagai penyebab thalassemia-αo
dan thalassemia-α+
di seluruh dunia (Huisman et al., 1997).10
Mutasi paling umum pada populasi Asia–Pasifik adalah delesi 4,2 Kb dan 3,7 Kb yang menyebabkan thalassemia-α+
(Hill et al., 1989). Mutasi paling sering di Populasi Asia Tenggara yang mendapat pengaruh kuat unggun gen Mongoloid adalah mutasi –SEA
sepanjang 17,5 Kb pada gen globin-α1
maupun globin-α2 (Bowden et al., 1992).
Mutan ini menyebabkan sintesis protein globin-α gagal karena tidak terbentuknya mRNA, sehingga bentuk homozigotnya menyebabkan bayi Hydrop Fetalis yang bersifat lethal (Pressley et al., 1980; Wasi, 1983).12,13
Distribusi mutan tersebar pada populasi Thailand, Malaysia dan Philipina dengan frekuensi polymorfik mencapai 5% (Wasi, 1981).14
Pengetahuan tentang dasar molekular thalassemia-α sangat penting untuk mempersiapkan prenatal diagnosis pada awal kehamilan, yang merupakan salah satu strategi untuk mengurangi insidensi penderita thalassemia-α yang baru. Sampai saat ini belum ada terapi kuratif yang memadai untuk penderita, walaupun cangkok sumsum tulang telah berhasil dilakukan, tetapi dibutuhkan
biaya yang sangat besar dan survival ratenya maksimal 15 tahun (Kanokpongsakdi et al., 1990; Cao et al., 1999).15,16
Terapi gen sendiri untuk penderita thalassemia-α memang telah dilakukan di Amerika tetapi masih terbatas pada skala penelitian belum untuk pelayanan (Weatherall and Clegg, 2001).17
Dengan demikian dapat disimpulkan bahwa tindakan preventif merupakan strategi yang paling tepat dalam managemen penyakit thalassemia.
Berdasarkan latar belakang di atas maka dilakukan penelitian tentang kajian molekular thalassemia-α pada penduduk kota Medan untuk mengetahui keberadaan mutasi paling umum del –SEA
pada berbagai kelompok suku di Medan. Data yang diperoleh diharapkan dapat menjadi acuan dasar dalam pengembangan prenatal diagnosis dalam managemen penyakit thalassemia di Medan dan sekitarnya sehingga munculnya bayi hidrop fetalis dan Hb H dapat dihindari.
BAHAN DAN CARA PENELITIAN Populasi dan Sampel
Sampel DNA dikoleksi dari darah vena 1.521 individu dewasa sehat, pendonor darah dengan kisaran umur 18–59 tahun, terdiri dari 1.306 laki-laki dan 215 perempuan. Sampel darah dikoleksi dari 7 kelompok etnik penduduk kota Medan yang mewakili populasi penduduk yang dominan. Komposisi jumlah sampel wakil tiap suku diambil sedemikian rupa sehingga mendekati keadaan sebenarnya yang merupakan representasi komposisi penduduk kota Medan berdasarkan data Sensus Penduduk tahun 2000 (Katalog BPS; 2110.12.).18
Cara Penelitian
Terhadap 51 sampel darah dari keseluruhan 1.521 sampel yang diperiksa, yang terdeteksi sebagai carrier thalasemia-α, secara hematologis atau biokimiawi berdasarkan serangkaian hasil pemeriksaan indeks hematologis, serum feritin dan tranferin, kuantifikasi HbA2 dan
keberadaan badan inklusi, selanjutnya dilakukan pemeriksaan DNA.
Isolasi DNA dari buffycoat dilakukan dengan metode modifikasi Lysis buffer Sucrose–Tris HCl-SDS (Gibco-BRL) dilanjutkan dengan purifikasi DNA menggunakan Proteinase-K (Merck) dan Proteinase-E (Merck). Deteksi mutan –SEA
Tabel 1. Distribusi jumlah sampel penelitian tiap suku terhadap komposisi penduduk Kota Medan pada Sensus Penduduk 2000 dan jumlah carrier Thalassemia-α.
* SP = Sensus Penduduk
A4 Forward = 5’-GGG-GCG-
CCT-TGG-GGA-GGT-TC-3’
A1B Reverse =
5’-GTT-CCC-TGA-GCC-CCG-ACA-CG-3’
A9 Reverse =
5’-ATA-TAT-GGG-TCT-GGA-AGT-GTA-TC-3’
Reaksi dilakukan dengan komposisi 10x buffer PCR (Roche) 2 mM dNTPs (Perkin-Elmer), 25 mM MgCl2, 10 pmole
masing-masing primer dan 0,2 IU enzim Tag
Polymerase (Perkin Elmer) dengan
Termocycler Gene Amp®
PCR System 2400 Perkin Elmer. Siklus PCR diawali dengan hot start pada 95o
C selama 1 menit, annealing pada suhu 63o
C selama 1 menit dikuti dengan fase ekstensi selama 30 detik pada suhu 72o
C. Inkubasi utama dilakukan pada siklus denaturasi pada suhu 95o
C selama 1 menit; annealing pada suhu 63o
C selama 1 menit dan ekstensi pada suhu 72o
C selama 1 menit sebanyak 30 siklus. Diakhiri dengan fase terminasi dengan suhu denaturasi 95o
C selama 30 detik; annealing pada 63o
C selama 1 menit dan ekstensi pada suhu 72o
C selama 10 menit. Pasangan primer A4 dan A1B akan
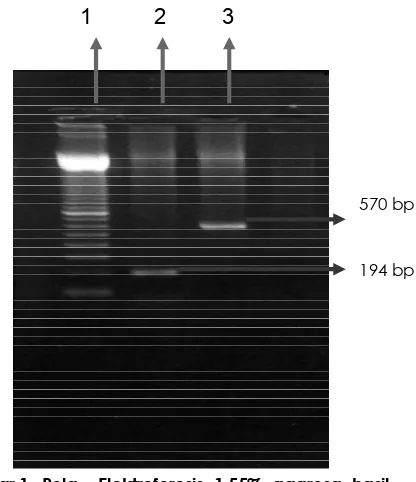
menghasilkan fragmen DNA sepanjang 194 bp pada sampel DNA normal maupun mutan–SEA
, sedangkan pasangan primer A4
dengan A9 akan menghasilkan fragment DNA
sepanjang 570 bp pada sampel DNA mutan–
SEA
dan tidak dijumpai pada DNA normal. Visualisasi fragment DNA hasil PCR dilakukan dengan elektroforesis1,55% agarosa
dalam buffer TBE 1x (Gibco-BRL) dengan pengecatan ethidium bromida. Sebagai standard digunakan DNA marker DNA 100bp ladder (Amersham Pharmacia Biotech).
HASIL PENELITIAN
Pola elektroforesis hasil PCR DNA mutan del –SEA
17 Kb Southeast Asia type dan pola DNA normal atau non-del-SEA
pada 1,55% agarosa dapat dilihat pada gambar berikut:
Gambar 1. Pola Elektroforesis 1,55% agarosa hasil PCR sampel DNA del --SEA . 1= Marker DNA 100 bp Ladder, 2 = DNA non-del-SEA, 3 = DNA del-SEA
Jumlah Sampel Penelitian
Jumlah Penduduk
Medan SP 2000* Ratio ∑ : SP
∑ Carrier Thal-α
Suku n % % n %
1. Batak 503 33,07 641.782 33,70 Relatif sama 9 1,78
2. Jawa 466 30,64 628.898 33,03 Relatif sama 17 3,64
3. Cina 223 14,66 202.839 10,65 Relatif sama 15 6,72
4. Melayu 136 8,94 125.557 6,59 Relatif sama 6 4,41
5. Minangkabau 128 8,42 163.774 8,60 Relatif sama 2 1,56
6. Aceh 57 3,75 53.011 2,78 Relatif sama 2 3,07
7. Nias 8 0,53 13,159 0,69 Relatif sama 0 0
8. Lain-lain suku 0 0,00 75.253 3,95 Berbeda 0 0
Jumlah 1.521 100 1.904.273 100 51 3,35
1 2 3
570 bp
Tabel 2. Distribusi Carrier Thalassemia-α0 del –SEA 17,5 Kb dan non- del--SEA 17,5 Kb pada berbagai suku penduduk Kota Medan
∑ Sampel Penelitian
∑carrier thal-α
∑carrier
Del-SEA 17,5 kb
∑carrier Non Del-SEA 17,5 Kb
Suku n % n % n % n %
1. Batak 503 33,07 9 1,78 6 1,19 3 0,59
2. Jawa 466 30,64 17 3,64 8 1,71 9 1,93
3. Cina 223 14,66 15 6,72 6 2,69 9 4,03
4. Melayu 136 8,94 6 4,41 4 2,94 2 1,47
5. Minangkabau 128 8,42 2 1,56 0 0,00 2 1,56
6. Aceh 57 3,75 2 3,07 2 3,50 0 0,00
7. Nias 8 0,53 0 0 0 0,00 0 0,00
Jumlah 1.521 100 51 3,35 26 1,70 25 1,64
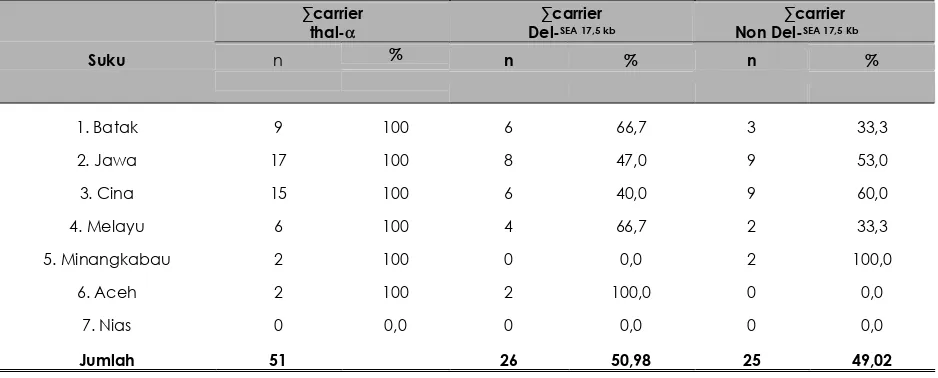
Tabel 3. Persentase carrier thal-α del-SEA terhadap seluruh carrier thal-α pada bebagai suku di Medan
∑carrier thal-α
∑carrier
Del-SEA 17,5 kb
∑carrier Non Del-SEA 17,5 Kb
Suku n % n % n %
1. Batak 9 100 6 66,7 3 33,3
2. Jawa 17 100 8 47,0 9 53,0
3. Cina 15 100 6 40,0 9 60,0
4. Melayu 6 100 4 66,7 2 33,3
5. Minangkabau 2 100 0 0,0 2 100,0
6. Aceh 2 100 2 100,0 0 0,0
7. Nias 0 0,0 0 0,0 0 0,0
Jumlah 51 26 50,98 25 49,02
Deteksi mutasi gen delesi –SEA
17,5 Kb terhadap 51 sampel DNA carrier
thalassemia-α pada penduduk kota Medan menunjukkan hasil seperti yang diperlihatkan pada Tabel 2.
PEMBAHASAN
Analisis molekular terhadap 51 sampel DNA yang dicurigai sebagai pembawa sifat (carrier) thalassemia-α, 26 sampel (51%) di antaranya ternyata positif mengalami mutasi delesi del-SEA
sepanjang 17,5 Kb pada kromosom 11. Hasil penelitian ini tidak mengejutkan karena sebelumnya telah dilaporkan bahwa mutasi del-SEA
merupakan mutasi utama pada populasi Asia Tenggara dan Pasifik di samping mutasi delesi utama lainnya seperti del (α)4,2
dan del (α)3,7
dan mutasi minor delesi –Fil
, --Thai
dan –HW
yang
sering dijumpai pada populasi Filipina, Thailand dan Hawai (Wasi, 1983).13
Mutasi –SEA
diakibatkan delesi sepanjang kira-kira 17,5 Kb yang mengakibatkan sebagian fragmen kedua gen globin-α, baik α1
maupun α2 ikut hilang, sehingga sintesis globin-α tidak terjadi atau thalassemia-α0
(Liebhaber et al., 1985).21
Mutan ini banyak dijumpai pada populasi yang mendapat kuat unggun gen Mongoloid misalnya Asia Timur maupun Asia Tenggara. Keberadaan gen ini dalam suatu populasi perlu diwaspadai karena bentuk homozigotnya menghasilkan bayi hydrop fetalis yang bersifat lethal. Jika gen tersebut berkombinasi dengan alel lainnya yang juga sering dijumpai pada populasi Asia Tenggara seperti del (α)4,2
, del (α)3,7
sama dengan thalassemia Intermedia atau Mayor yang membutuhkan transfusi darah rutin seumur hidupnya (Bowden et al., 1997).11
Hasil penelitian menunjukkan bahwa mutan del–SEA
dominan (>50%) ditemukan pada total carrier thalassemia-α dibanding dengan mutan lainnya pada populasi Batak dan Melayu (66,7%), dan Aceh (100%). Mutan ini ditemukan dengan frekuensi lebih rendah pada populasi Medan non-Melayu yaitu 47% dan 40% pada populasi Jawa dan China. Angka tersebut relevan dengan hasil penelitian sebelumnya. George (1994) melaporkan bahwa frekuensi gen del–SEA
pada populasi Melayu di Semenanjung Malaya mencapai 4,5% atau 61,3% dari keseluruhan kasus thalassemia-α.22
Laporan lainnya juga menyebutkan bahwa mutasi yang sering dijumpai pada populasi China adalah del (α)((Lie et al., 1982; Yang et al, 1985).5,6
Dan pada populasi Jawa del (α)4,2
, del (α)3,7
(Setianingsih et al., 2003).8
Jika dilihat dari frekuensi gen dalam populasi secara keseluruhan maka mutant ini paling banyak dijumpai berturut-turut pada populasi Aceh (3.5%); Melayu (2.97%); China (2,69%); Jawa (1.71%) dan Batak (1,19%) dan tidak dijumpai pada populasi Minangkabau and Nias.
Hasil penelitian ini sangat berguna dalam pengembangan prenatal diagnosis untuk populasi Medan, dengan menetapkan mutasi del–SEA
menjadi prioritas utama dalam skrining prenatal diagnosis pada pasangan pembawa sifat thalassemia-α, terutama jika mereka berasal dari etnik Batak, Melayu, Aceh, Jawa, dan China.
KESIMPULAN
Dari hasil penelitian dapat disimpulkan bahwa:
- Mutant thalassemia-α paling sering dijumpai pada populasi Medan adalah del -SEA
- Mutan del-SEA dijumpai terutama pada suku Aceh, Batak dan Jawa melebihi 50% dari total kasus thalassemia-αyang ada.
- Hasil penelitian dapat dilakukan sebagai acuan untuk pengembangan Prenatal Diagnosis thalassemia-α untuk populasi Medan dengan menetapkan mutan del-SEA
sebagai prioritas utama deteksi.
DAFTAR PUSTAKA
1. Flint J, Harding R, Clegg JB and Boyce A (1993). Why are some genetic diseases so common? Distinguishing selection from other process by molecular analysis of globin gene variants. Hum Genet. 91:91-117.
2. Hill, A.V.S., O’Shaughnessy, D.F. and Clegg, J.B. 1989. Haemoglobin and globin gene variants in the Pacific. In: Hill A.V.S. and Serjeantson S.W. (eds); The Colonization of the Pacific; a genetic trial. Clarendon Press. Oxford. UK.
3. WHO (1994) Guidelines for the control of haemoglobin disorders. Report of the VIth annual Meeting of the WHO Working Group on Haemoglobinopathies. Cagliari. Sardinia, 8-9 April 1989. Word Health Organization, Geneva.
4. Higgs D.R, Wood WG., Jarman A.P., Vickers M.A., Wilkie A.O., Lamb J., Vyas P and Bannett J.P. (1990) The alpha thalassenias. Ann.N.Y. Acad.Sci. 612:15-22.
5. Li, AMC, Lee, FT and Tood D (1982) The screening of Chinese blood cord blood for hemoglobinopathies. Hum Hered 32 : 62-65
6. Yang TY, Yang XY and Chen WC (1985) Thalassemia in China. Ann N.Y. Acad. Sci 445: 92-97.
7. Wong H.B (1983) Thalassemia as community health in Southeast Asia. Naskah Lengkap Kongres National PHDTI. Yogyakarta 24-26 September 1983.
8. Setianingsih, I., Harahap, A. and Nainggolan, I.M. 2003. Alpha thalassaemia in Indonesia: phenotypes and molecular defects. Tropical Diseases, Edited by Marzuki, Verhoef, and Snippe. Kluwer Academic/Plenum Publishers, New York. 47-56.
9. Hariman, H., Abdullah, I., Nasution, B. 1984. Some aspects of alpha-thalassamias in the region. MedikaX(1). 54: 24-28.
10. Huisman, T.H.J., Carver, M.F.H. and Baysal, E. 1997. A Syllabus of
Thalassemia Mutations. The Sickle Cell
11.Bowden, D.K., Vickers, M.A. and Higgs, D.R. 1992. A PCR-based strategy to detect the common severe determinants of α-thalassemia. Br. J. Haematol. 81: 104-108.
12. Pressley, L., Higgs, D.R., Clegg, J.B. and Weatherall, D.J. 1980. Gene deletions in an α thalassemia prove that the 5’ξ locus is functional. Proc. Natl. Acad. Sci. USA. 77: 3586-3589.
13. Wasi, P. 1983. Population Screening. In: Weatherall, D.J. (ed). Method in Hematology. The Thalassemias. London. Churchill Livingstone. 134-144.
14. Wasi, P. 1981. Haemoglobinopathies including thalassemia. Clin. Haematol. 10: 707-729.
15. Kanokpongsakdi, S., Winichagoon, P. and Fucharoen, S. 1990. Control of thalassaemia in Southeast Asia. Journal of Paediatrics, Obstetrics and Gynaecology. Thailand. 9-14.
16. Cao, A., Galanello, R. and Rosatelli, M.C. 1999. Prenatal diagnosis and screening of the haemaglobinopathies. The Electronic Journal Of the International Federation Of Clinical Chemistry And Laboratory Medicine. Italy. 3: 1-11.
17. Weatherall DJ and Clegg JB (2001) The Thalassemia Syndromes, 4th
eds. Blackwell Scientific Publ. Oxford. P 31-32
18. Badan Pusat Statistik Propinsi Sumatera Utara. 2001. Angka Sementara Penduduk Sumatera Utara : Hasil Sensus Penduduk 2000. Katalog BPS: 2110.12.
19. Nicholls, R.D., Fischel-Ghodsian, N., Higgs, D.R. 1987. Recombination at the human α-globin gene cluster: sequence features and topological constraints. Cell. 49: 369-374.
20. Liebhaber S.A, Goorsens M and Kan Y.W (1981) Cloning ang complete nucleotide sequence of the human 5’α-globin gene. Proc. Natl.Acad.Scid USA 77:7054-7063. 21. Chang, J.G., L.S., Lin, C.P and Chen C.P.
1991. Rapid diagnosis of -thalassaemia-1 of Southeast Asia type and hydrops fetalis by polymerase chain reaction. Blood. 78: 853-854