IDENTIFIKASI DNA BAKTERI MENGGUNAKAN METODE
EKSTRAKSI CIRI SPACED K-MERS DENGAN JARINGAN
SYARAF TIRUAN SEBAGAI CLASSIFIER
CUT MALISA IRWAN
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
PERNYATAAN MENGENAI SKRIPSI DAN
SUMBER INFORMASI SERTA PELIMPAHAN HAK CIPTA
Dengan ini saya menyatakan bahwa skripsi berjudul Identifikasi DNA Bakteri Menggunakan Metode Ekstraksi Ciri Spaced K-Mers dengan Jaringan Syaraf Tiruan sebagai Classifier adalah benar karya saya dengan arahan dari komisi pembimbing dan belum diajukan dalam bentuk apa pun kepada perguruan tinggi mana pun. Sumber informasi yang berasal atau dikutip dari karya yang diterbitkan maupun tidak diterbitkan dari penulis lain telah disebutkan dalam teks dan dicantumkan dalam Daftar Pustaka di bagian akhir skripsi ini.
Dengan ini saya melimpahkan hak cipta dari karya tulis saya kepada Institut Pertanian Bogor.
Bogor, Juli 2013
Cut Malisa Irwan
ABSTRAK
CUT MALISA IRWAN. Identifikasi DNA Bakteri Menggunakan Metode Ekstraksi Ciri Spaced K-Mers dengan Jaringan Syaraf Tiruan sebagai Classifier. Dibimbing oleh TOTO HARYANTO dan HABIB RIJZAANI.
Ekstraksi ciri adalah proses pengambilan penciri dari suatu objek yang dapat menggambarkan karakteristik dari objek tersebut. Pada penelitian ini, metode ekstraksi ciri yang digunakan adalah spaced k-mers. Metode ekstraksi ciri tersebut digunakan untuk mengambil penciri sekuens DNA dari tiga genus, yaitu: Bacillus,
Burkholderia, dan Pseudomonas. Jaringan syaraf tiruan digunakan untuk menganalisis data biologi molekuler tiga genus. Pada penelitian ini, metode ekstraksi ciri spaced k-mers menggunakan nilai parameter w = 3, dan d = 0, 1, 2, serta panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp. Nilai sensitivity dan
specificity terbaik diperoleh untuk known organisms pada panjang fragmen 1 Kbp, yaitu dengan nilai sensitivity 0.9716 dan nilai specificity 0.9854.
Kata kunci: jaringan syaraf tiruan, sensitivity, sekuens DNA, spaced k-mers,
specificity
ABSTRACT
CUT MALISA IRWAN. Identification Bacteri DNA Using Feature Extraction Spaced K-Mers with Artifical Neural Network as Classifier. Supervised by TOTO HARYANTO and HABIB RIJZAANI.
Feature extraction is the process of taking an object identifiers that describes it’s characteristics. In this study, spaced k-mers feature extraction method was employed. This method was used to retrieve the data identifier of DNA sequence of the three genus, namely Bacillus, Burkholderia, and Pseudomonas. Artificial neural network was used to analyze molecular biology data from the three genus. The feature extraction methods uses the following setup: w = 3, and d = 0, 1, 2 and fragment length 100 bp, 400 bp, 800 bp, and 1 Kbp. The best sensitivity and the best specificity were achieved for known organisms at 1 Kbp fragment length with value 0.9716 and 0.9854, respectively.
Skripsi
sebagai salah satu syarat untuk memperoleh gelar Sarjana Komputer
pada
Departemen Ilmu Komputer
IDENTIFIKASI DNA BAKTERI MENGGUNAKAN METODE
EKSTRAKSI CIRI SPACED K-MERS DENGAN JARINGAN
SYARAF TIRUAN SEBAGAI CLASSIFIER
CUT MALISA IRWAN
DEPARTEMEN ILMU KOMPUTER
FAKULTAS MATEMATIKA DAN ILMU PENGETAHUAN ALAM INSTITUT PERTANIAN BOGOR
Judul Skripsi : Identifikasi DNA Bakteri Menggunakan Metode Ekstraksi Ciri
Spaced K-Mers dengan Jaringan Syaraf Tiruan sebagai Classifier
Nama : Cut Malisa Irwan
NIM : G64080007
Disetujui oleh
Toto Haryanto, SKom MSi Pembimbing I
Habib Rijzaani, MSi Pembimbing II
Diketahui oleh
Dr Ir Agus Buono, MSi MKom Ketua Departemen
PRAKATA
Puji syukur penulis panjatkan kepada Allah Subhanahu wa-ta'ala atas segala rahmat dan karunia-Nya sehingga penulis dapat menyelesaikan skripsi yang berjudul “Identifikasi DNA Bakteri Menggunakan Metode Ekstraksi Ciri
Spaced K-Mers dengan Jaringan Syaraf Tiruan sebagai Classifier”. Penulisan skripsi ini tak lepas dari bantuan banyak pihak. Oleh karena itu, penulis ingin menyampaikan rasa terima kasih kepada:
• Kedua orang tua penulis, Bapak Teuku Irwan dan Ibu Nani Erwani, atas pola pendidikan luar biasa yang telah diberikan kepada penulis.
• Bapak Toto Haryanto SKom MSi dan Bapak Habib Rijzaani MSi selaku dosen pembimbing skripsi. Terima kasih atas segala ilmu, bantuan, serta nasehat-nasehat yang diberikan kepada penulis.
• Bapak Dr Wisnu Ananta Kusuma ST MT selaku dosen penguji.
• Saudara Dony Satria, atas segala motivasi, semangat, dukungan, masukan, dan saran selama proses pengerjaan skripsi ini.
• Seluruh rekan-rekan dari Departemen Ilmu Komputer, atas segala masukan dan saran selama proses pengerjaan skripsi ini.
Semoga karya ilmiah ini bisa memberikan manfaat untuk perkembangan dunia teknologi informasi dan pertanian di Indonesia.
Bogor, Juli 2013
DAFTAR ISI
DAFTAR TABEL vi
DAFTAR GAMBAR vi
DAFTAR LAMPIRAN vi
PENDAHULUAN 1
Latar Belakang 1
Tujuan Penelitian 2
Ruang Lingkup Penelitian 2
Manfaat Penelitian 2
METODE PENELITIAN 2
Studi Literatur 3
Pengumpulan Data 3
Praproses 4
Ekstraksi Ciri SpacedK-Mers 5
K-Fold Cross Validation 7
Klasifikasi Jaringan Syaraf Tiruan (JST) 8
Pengujian 10
Analisis 10
HASIL DAN PEMBAHASAN 12
Praproses Data 12
Ekstraksi Ciri SpacedK-Mers 13
5-Fold Cross Validation 13
Klasifikasi JST 14
Pengujian 15
Analisis Hasil 15
SIMPULAN DAN SARAN 20
Simpulan 20
Saran 21
DAFTAR PUSTAKA 21
LAMPIRAN 22
DAFTAR TABEL
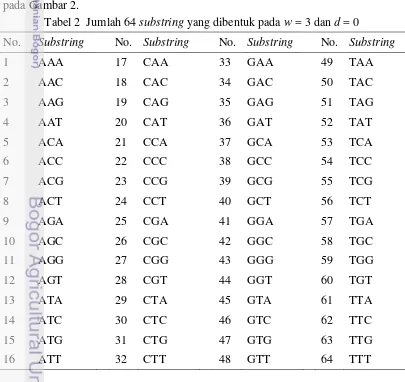
1 Pola untuk spacedk-mers dengan nilai w = 3 dan d = 0, 1, 2 5 2 Jumlah 64 substring yang dibentuk pada w = 3 dan d = 0 6
3 Proses pada metode 5-fold cross validation 8
4 Parameter pelatihan JST menggunakan back propagation 9
5 Confusion matrix genus 1 11
6 Tiga genus data sekuens DNA dengan panjang fragmen 100 bp 12 7 Tiga genus data sekuens DNA dengan panjang fragmen 400 bp 12 8 Tiga genus data sekuens DNA dengan panjang fragmen 800 bp 12 9 Tiga genus data sekuens DNA dengan panjang fragmen 1 Kbp 13 10 Jumlah data latih dan data uji dari ketiga genus 13 11 Nilai MSE dari proses pelatihan pada data latih 14 12 Nilai sensitivity dan specificity untuk known organisms dari setiap genus 16 13 Nilai sensitivity dan specificity untuk new organisms dari setiap genus 17 14 Nilai rata-rata sensitivity dan specificity untuk known organisms dari
ketiga jenis genus 19
15 Nilai rata-rata sensitivity dan specificity untuk new organisms dari ketiga
jenis genus 19
DAFTAR GAMBAR
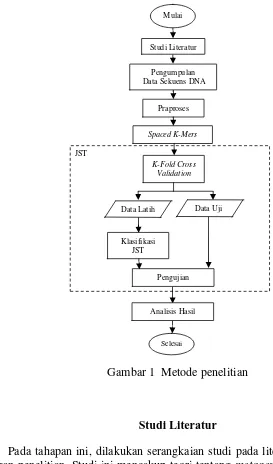
1 Metode penelitian 3
2 Ilustrasi dari proses slidingwindow 7
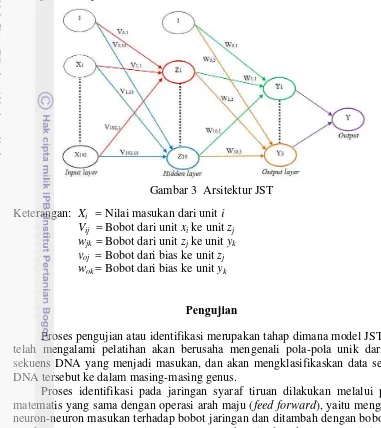
3 Arsitektur JST 10
4 Grafik nilai sensitivity untuk known organisms setiap genus berdasarkan
panjang fragmen 16
5 Grafik nilai specificity untuk known organisms setiap genus berdasarkan
panjang fragmen 17
6 Grafik nilai sensitivity untuk new organisms setiap genus berdasarkan
panjang fragmen 18
7 Grafik nilai specificity untuk new organisms setiap genus berdasarkan
panjang fragmen 18
8 Grafik nilai rata-rata sensitivity dari ketiga genus 19 9 Grafik nilai rata-rata specificity dari ketiga genus 20
DAFTAR LAMPIRAN
1 Daftar organisme untuk known organisms untuk setiap genus dengan panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp 22 2 Daftar organisme untuk new organisms untuk setiap genus dengan
panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp 24
3 Algoritme pelatihan JST back propagation 25
5 Confusion matrix untuk know organisms dari setiap genus dengan panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp 28 6 Confusion matrix untuk new organisms darisetiap genus dengan panjang
PENDAHULUAN
Latar Belakang
Deoxyribo nucleic acid (DNA) adalah sejenis asam nukleat yang tergolong dalam biologi molekuler utama penyusun setiap organisme. DNA mengandung tiga komponen: deoxyribo (gula dengan 5 karbon), urutan dari fosfat, dan 4 basa nitrogen yaitu adenine (A), thymine (T), guanine (G), dan cytosine (C). DNA berfungsi untuk menyimpan informasi genetik pada suatu organisme. DNA pada setiap spesies akan berbeda satu sama lainnya. Adanya perbedaan genetik di antara individu atau organisme ini, melahirkan berbagai sistem identifikasi berbasis DNA. Bioinformatika merupakan salah satu ilmu yang mempelajari penerapan teknik komputasi untuk mengidentifikasi dan menganalisis informasi biologis, terutama dengan menggunakan sekuens DNA dan asam amino serta informasi yang berkaitan dengannya. Pelacak spesifik gen dapat dikembangkan dengan memanfaatkan kemajuan bioinformatika pada teknik-teknik biologi molekuler.
Metagenome melibatkan suatu teknik yang secara khusus ditunjukkan untuk mengumpulkan gen-gen secara langsung dari suatu lingkungan, diikuti dengan menganalisis informasi genetika yang terkandung di dalamnya (Riesenfeld et al. 2004). Data yang digunakan pada saat proses pembacaan metagenome dapat berupa data DNA yang diperoleh dari lingkungan, sehingga terdapat kemungkinan bahwa hasil pembacaan tersebut merupakan percampuran beberapa fragmen dari organisme yang berbeda. Oleh sebab itu, fragmen yang saling bercampur ini bisa mengakibatkan kesalahan pengklasifikasian. Untuk mengatasi permasalahan ini, diperlukan suatu metode ekstraksi ciri dan metode klasifikasi untuk menentukan jenis organisme atau tingkatan taksonomi dari suatu fragmen metagenome
(Wooley et al. 2010). Proses klasifikasi tersebut dapat dilakukan dengan menggunakan ciri-ciri biokimia, misalnya jenis-jenis DNA, jenis-jenis protein, dan jenis-jenis enzim, sehingga dapat menentukan hubungan kekerabatan antara makhluk hidup satu dengan lainnya.
Salah satu metode ekstraksi ciri yang dapat digunakan untuk melakukan klasifikasi sekuens DNA adalah metode k-mers. Penelitian menggunakan k-mers
telah dilakukan, di antaranya oleh McHardy et al. (2007), yang telah melakukan penelitian klasifikasi terhadap 340 organisme menggunakan metode ekstraksi ciri
k-mers dan metode klasifikasi support vector machine (SVM). Hasil akurasi yang didapat dari penelitian ini untuk panjang fragmen ≥ 5 Kbp, yaitu berkisar antara 60% sampai lebih dari 90% di setiap tingkat takson, sedangkan akurasi untuk takson genus dan order terus menurun dengan signifikan pada panjang fragmen ≤ 3 Kbp. Akurasi tersebut turun mulai dari 40% untuk panjang fragmen 3 Kbp hingga < 10% untuk panjang fragmen 1 Kbp.
2
Berdasarkan pemaparan latar belakang sebelumnya, pada penelitian ini penulis akan mencoba melakukan identifikasi pola sekuens DNA (fragmen
metagenome) bakteri dari genus Bacillus, Burkholderia, dan Pseudomonas
menggunakan metode ekstraksi ciri spaced k-mers dengan jaringan syaraf tiruan (JST) sebagai classifier.
Tujuan Penelitian
Penelitian ini bertujuan untuk membuat model klasifikasi berbasis JST yang diimplementasikan untuk melakukan identifikasi sekuens DNA terhadap tiga jenis genus bakteri, yaitu genus Bacillus, Burkholderia, dan Pseudomonas, dengan menggunakan spacedk-mers sebagai metode ekstraksi ciri.
Ruang Lingkup Penelitian
Ruang lingkup penelitian ini meliputi:
1 Data sekuens DNA terdiri atas 3 genus, yaitu Bacillus, Burkholderia, dan
Pseudomonas dengan panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp. 2 Data latih dan data uji sekuens DNA dari known organisms terdiri atas 50
organisme.
3 Data uji sekuens DNA dari new organisms terdiri atas 30 organisme.
4 Data sekuens DNA dari 3 genus tersebut memiliki format penyimpanan .fna.
Manfaat Penelitian
Manfaat penelitian ini ada dua, yaitu:
1 Dapat melakukan identifikasi sekuens DNA bakteri genus Bacillus,
Burkholderia, dan Pseudomonas dengan menggunakan metode ekstraksi ciri
spacedk-mers dan metode JST sebagai classifier.
2 Mengetahui tingkat akurasi pengklasifikasian sekuens DNA bakteri genus
Bacillus, Burkholderia, dan Pseudomonas dengan menggunakan metode ekstraksi ciri spacedk-mers dan metode JST sebagai classifier.
METODE PENELITIAN
3
Gambar 1 Metode penelitian
Studi Literatur
Pada tahapan ini, dilakukan serangkaian studi pada literatur yang berkaitan dengan penelitian. Studi ini mencakup teori tentang metagenome, bioinformatika, sekuens DNA, spacedk-mers, JST, dan sebagainya.
Pengumpulan Data
Data yang digunakan dalam penelitian ini merupakan data sekuens DNA diperoleh dari National Center for Biotechnology Information (NCBI) pada situs ftp://ftp.ncbi.nih.gov/genomes/Bacteria/all.fna.tar.gz. NCBI merupakan suatu institusi yang fokus sebagai sumber informasi perkembangan biologi molekuler. Kegiatan yang dilakukan oleh NCBI di antaranya adalah membuat database yang dapat diakses oleh publik, melakukan riset biologi terkomputasi, mengembangkan
software penganalisis data genome, dan menyebarkan informasi biomedical yang kesemuanya diharapkan mengarah pada pemahaman yang lebih baik tentang proses-proses molekuler yang mempengaruhi manusia dan kesehatannya.
Mulai
Pengumpulan Data Sekuens DNA
Praproses
Spaced K-Mers
K-Fold Cross Validation
Data Latih
Klasifikasi JST
Pengujian Studi Literatur
Data Uji
4
Setelah didapatkan data sekuens DNA dari taksonomi NCBI, selanjutnya data tersebut akan diproses pada software MetaSim (version 0.9.1). MetaSim merupakan suatu perangkat lunak simulasi yang dapat digunakan untuk menghasilkan data metagenome (Richter et al. 2009). MetaSim melakukan pencarian data sesuai dengan parameter yang dimasukan oleh pengguna. Pada penelitian ini, parameter yang ditentukan ada dua, yaitu number of reads dan
mean. Number of reads merupakan jumlah sekuens DNA yang diinginkan oleh pengguna, sedangkan mean adalah panjang fragmen dari sekuens DNA.
Pada penelitian ini, yang dimaksud dengan known organisms adalah data uji yang diperoleh dari dataset yang telah diketahui jenis organismenya yang dihasilkan melalui tahapan k-fold cross validation, sedangkan new organisms
merupakan kumpulan jenis organisme berbeda dari known organisms yang dibangkitkan melalui software MetaSim, tetapi termasuk ke dalam genus yang sama dengan known organisms, yaitu: Bacillus, Burkholderia, dan Pseudomonas. Nilai number of reads yang digunakan pada penelitian ini untuk data latih dan data uji pada known organisms adalah 1800, 1735, 1790, dan 1790, sedangkan nilai number of reads untuk data uji pada new organisms adalah 360, 347, 358, dan 358. Nilai mean yang digunakan pada penelitian ini untuk known organisms
dan new organisms adalah 100 bp, 400 bp, 800 bp, dan 1 Kbp. Daftar organisme untuk known organisms dan new organisms dapat dilihat pada Lampiran 1 dan Lampiran 2. Keluaran dari pengolahan MetaSim ini adalah FastA. FastA merupakan file yang berisi sekuens DNA yang sudah terfragmen sesuai dengan nilai parameter yang dimasukan oleh pengguna.
Data metagenome hasil simulasi dari MetaSim yang akan digunakan pada penelitian ini merupakan data sekuens DNA yang terdiri atas seri huruf yang mewakili struktur primer dari molekul DNA, yaitu huruf A, C, G, dan T. Data tersebut merupakan data sekuens DNA bakteri pada known organisms dan new organisms yang terdiri atas 3 genus, yaitu Bacillus, Burkholderia, dan
Pseudomonas dengan panjang fragmen yaitu 100 bp, 400 bp, 800 bp, dan 1 Kbp.
Praproses
Pada tahapan praproses akan dilakukan proses parsing, yaitu proses pemisahan antara informasi sources dan informasi sekuens DNA, sehingga hanya informasi sekuens DNA yang akan menjadi ciri dari sebuah organisme.
Memisahkan sources:
>r30.1|SOURCES={GI=50196905,fw,33345803334680}|ERRORS={}|SOURC_ 1="Bacillus anthracis str. 'Ames Ancestor'"
(2b301d2cec11c944b70447bada91610998f9ea15) Sekuens DNA hasil parsing:
5 Ekstraksi Ciri Spaced K-Mers
Ekstraksi ciri adalah proses pengambilan penciri yang terdapat pada suatu citra atau suatu data. Ekstraksi ciri diklasifikasikan ke dalam tiga tingkat yaitu
low-level, middle-level, dan high-level. Low-level feature merupakan ekstraksi ciri berdasarkan isi visual seperti warna dan tekstur, middle-level feature merupakan ekstraksi ciri setiap objek dalam citra atau data dan mencari keterhubungan di antara objek tersebut, sedangkan high-level feature merupakan ekstraksi ciri berdasarkan informasi semantik yang terkandung dalam citra atau data (Osadebey 2006).
Spaced k-mers merupakan sistem pemrosesan string, yang dapat digunakan untuk mengetahui intensitas atau banyaknya kemunculan substring tertentu, pada sebuah string. Intensitas kemunculan substring tersebut, dapat dijadikan sebagai penciri atau fitur dari suatu kelompok string. Hal tersebut merupakan landasan utama penggunaan spaced k-mers sebagai metode ekstraksi ciri pada penelitian ini, karena data yang akan digunakan dalam penelitian ini adalah data sekuens DNA yang merupakan data string.
Pada penelitian ini pola spaced k-mers yang akan digunakan yaitu w = 3 dan
d = 0, 1, dan 2. Mengacu pada penelitian Kusuma (2012), dikatakan bahwa pola yang akan menghasilkan akurasi tinggi dari klasifikasi adalah dengan nilai variabel w dan d tersebut. Variabel w (weight of pattern) adalah banyaknya basa nitrogen yang digunakan untuk membentuk sebuah pola, dan variabel d adalah jumlah don’t care. Nilai w pada spaced k-mers menunjukkan jumlah karakter yang diinginkan untuk membentuk sebuah substring (Kusuma 2012). Pola untuk
spacedk-mers dengan nilai w = 3 dan d = 0, 1, 2, adalah sebagai berikut:
ϖ
d=0,1,2w=3111 1*11 1**11
Jumlah kombinasi untuk metode spacedk-mers dengan nilai w = 3 dan d = 0, 1, dan 2 dapat dilihat pada Tabel 1. Metode ini akan memeriksa frekuensi nukleotida dari fragmen DNA mulai dari AAA sampai GGG, A*AA sampai G*GG, dan A**AA sampai G**GG. Sehingga akan didapat 192 kombinasi nukleotida. Pengertian dari simbol * (don’t care) pada fragmen DNA yang diperiksa adalah dapat berupa basa apapun, baik A, T, G, dan C, sedangkan untuk simbol ** berarti diperbolehkan pasangan basa manapun mengisi 2 bit tersebut. Sehingga kondisi ini dapat diisi oleh 24 pasang basa mulai dari AA, AC, AT, AG, dan seterusnya hingga GG.
Tabel 1 Pola untuk spacedk-mers dengan nilai w = 3 dan d = 0, 1, 2
w d Pola Jumlah
kombinasi
0 AAA, AAT, AAG, AAC,…, GGG 64
3 1 A*AA, A*AT, A*AG, A*AC,…, G*GG 64
2 A**AA, A**AT, A**AG, A**AC,…, G**GG 64
6
Misal, diketahui suatu stringS bernilai GGAATCCGA, dengan nilai w dan
d pada spacedk-mers adalah 3 dan 0.
S=GGAATCCGA
w=3, d 0
Pada string S, dapat dilihat bahwa karakter yang membentuk string tersebut ada empat, yaitu A, T, G, C dan nilai w dan d yang digunakan adalah w = 3 dan d = 0. Berdasarkan kedua informasi tersebut, dapat diketahui bahwa kemungkinan maksimal kombinasi substring yang dapat dibentuk adalah:
4×4 ×4=43 =64 substring
Kombinasi substring yang dibentuk pada metode spaced k-mers dengan w = 3 dan d = 0 dapat dilihat pada Tabel 2. Namun, 64 substring tersebut belum tentu muncul pada stringS. Cara mencari intensitas kemunculan substring tersebut pada
string S adalah dengan metode sliding window. Berdasarkan nilai k atau banyaknya karakter pada substring, ukuran sliding window yang digunakan adalah 3 karakter. Langkah kerja dari proses sliding window adalah, sliding window akan terus bergeser dari awal hingga akhir string S dengan jarak
overlapping sejauh 2 karakter. Ilustrasi dari proses sliding window dapat dilihat pada Gambar 2.
Tabel 2 Jumlah 64 substring yang dibentuk pada w = 3 dan d = 0
No. Substring No. Substring No. Substring No. Substring
1 AAA 17 CAA 33 GAA 49 TAA
2 AAC 18 CAC 34 GAC 50 TAC
3 AAG 19 CAG 35 GAG 51 TAG
4 AAT 20 CAT 36 GAT 52 TAT
5 ACA 21 CCA 37 GCA 53 TCA
6 ACC 22 CCC 38 GCC 54 TCC
7 ACG 23 CCG 39 GCG 55 TCG
8 ACT 24 CCT 40 GCT 56 TCT
9 AGA 25 CGA 41 GGA 57 TGA
10 AGC 26 CGC 42 GGC 58 TGC
11 AGG 27 CGG 43 GGG 59 TGG
12 AGT 28 CGT 44 GGT 60 TGT
13 ATA 29 CTA 45 GTA 61 TTA
14 ATC 30 CTC 46 GTC 62 TTC
15 ATG 31 CTG 47 GTG 63 TTG
7
Gambar 2 Ilustrasi dari proses slidingwindow
Dari Gambar 2, dapat diketahui substring apa saja yang dibentuk oleh string S beserta intensitas kemunculannya.
K-Fold Cross Validation
K-fold cross validation merupakan teknik yang membagi data ke dalam k
bagian untuk kemudian masing-masing bagian data tersebut akan dilakukan proses klasifikasi. Metode k-fold cross validation digunakan dengan tujuan agar akurasi yang dihasilkan pada penelitian ini merupakan akurasi secara umum, yang dapat merepresentasikan akurasi data secara keseluruhan. Langkah pertama dalam metode k-fold cross validation adalah menentukan nilai k. Nilai k adalah nilai yang menunjukkan jumlah pembagian data menjadi k-subset data. Pada penelitian ini, nilai k yang digunakan adalah 5 sehingga, metode k-fold cross validation yang digunakan pada penelitian menjadi 5-fold cross validation. Berdasarkan nilai k
tersebut, jumlah subset data yang dihasilkan adalah 5-subset data.
Setelah 5 subset data terbentuk, pilih sebuah subset data untuk dijadikan sebagai data uji. Selanjutnya, keempat subset data lain yang tidak terpilih dijadikan sebagai data latih. Proses pemilihan subset data uji dan subset data latih tersebut dilakukan secara berulang kali sehingga kelima subset data yang dihasilkan pernah menjadi subset data uji sebanyak tepat 1 kali.
Setiap kali diperoleh sebuah subset data uji, tahapan klasifikasi dapat dilakukan hingga diperoleh sebuah nilai akurasi klasifikasi. Berdasarkan jumlah
subset data yang digunakan, pada akhir dari penelitian ini akan dihasilkan 5 nilai akurasi klasifikasi. Kelima nilai akurasi tersebut akan dirata dan hasil rata-rata tersebut merupakan nilai akurasi klasifikasi akhir yang merepresentasikan nilai akurasi klasifikasi data secara keseluruhan. Proses pada metode 5-fold cross validation tersebut dapat dilihat pada Tabel 3.
8
Tabel 3 Proses pada metode 5-fold cross validation
Subset data uji Subset data latih Akurasi
Subset_1 Subset_2, Subset_3, Subset_4, dan Subset_5 Akurasi_1
Subset_2 Subset_1, Subset_3, Subset_4,dan Subset_5 Akurasi_2
Subset_3 Subset_1, Subset_2, Subset_4, dan Subset_5 Akurasi_3
Subset_4 Subset_1, Subset_2, Subset_3, dan Subset_5 Akurasi_4
Subset_5 Subset_1, Subset_2, Subset_3, dan Subset_4 Akurasi_5 Persamaan akurasi akhir klasifikasi adalah sebagai berikut:
Akurasi Akhir = ∑ Akurasii n
i=1
n ;n =5
Klasifikasi Jaringan Syaraf Tiruan (JST)
JST adalah sistem pemrosesan informasi yang memiliki karakter yang mirip dengan jaringan syaraf biologis, berupa generalisasi model matematika dari jaringan biologi yang didasarkan pada beberapa asumsi (Yani 2005). Proses pelatihan JST ditujukan agar model jaringan dapat mempelajari karakteristik dari setiap genus sehingga diperoleh suatu jaringan terbaik yang diharapkan mampu mendeteksi data sekuens DNA dengan akurat.
Sebelum melakukan pelatihan, dibutuhkan suatu matriks yang disebut dengan matriks target. Matriks target tersebut dibuat berdasarkan matriks data latih, yaitu matriks target digunakan untuk memberikan informasi kepada jaringan bahwa suatu kolom pada matriks data latih termasuk ke dalam genus pertama, genus kedua, atau genus ketiga. Dalam penelitian ini, genus pertama adalah
Bacillus, genus kedua adalah Burkholderia, genus ketiga adalah Pseudomonas. Karena JST membutuhkan matriks target dalam mempelajari karakteritik dari suatu genus, maka JST masuk ke dalam supervised learning.
Algoritme yang digunakan dalam tahap pelatihan JST adalah back propagation. Tahapan pelatihan JST menggunakan algoritme back propagation
9 Tabel 4 Parameter pelatihan JST menggunakan back propagation
Parameter Nilai
Inisialisasi bobot Nguyen-Widrow
Input layer 192 neuron
Hidden layer 10 neuron
Output layer 3 neuron
Fungsi aktivasi pada lapisan tersembunyi Sigmoid logaritmik Fungsi aktivasi pada lapisan output Sigmoid logaritmik Fungsi pelatihan jaringan Levenberg-Marquardt Fungsi pelatihan bobot Gradient descent momentum
Fungsi aktivasi yang digunakan pada penelitian ini adalah sigmoid logaritmik yang memiliki selang nilai (0,1). Persamaan dari fungsi aktivasi sigmoid logaritmik yaitu:
f x = 1 1+exp -x
Keterangan: f x = nilai output fungsi aktivasi
x = nilai input fungsi aktivasi
Pada penelitian ini, untuk pemilihan bobot dan bias awal pada tahapan pelatihan JST menggunakan metode Nguyen-Widrow. Pemilihan bobot awal sangat mempengaruhi JST dalam mencapai minimum global atau minimum lokal terhadap nilai error dan cepat tidaknya proses pelatihan menuju kekonvergenan. Algoritme inisialisasi Nguyen-Widrow dapat dilihat pada Lampiran 4.
Salah satu indikator yang digunakan untuk melihat baik atau tidaknya sebuah jaringan yang dihasilkan adalah nilai mean square error (MSE). MSE adalah rata-rata dari kesalahan pembelajaran jaringan (selisih antara ouput aktual dengan output target) yang dikuadratkan. Persamaan dari MSE adalah sebagai berikut:
MSE = ∑ tk-yk 2 n
k =1
n
Keterangan: tk = nilai output aktual ke-k
yk= nilai output target ke-k n = banyaknya nilai output
10
sehingga memberikan nilai MSE yang paling kecil. Arsitektur JST pada penelitian ini dapat dilihat pada Gambar 3.
Gambar 3 Arsitektur JST Keterangan: Xi = Nilai masukan dari unit i
Vij = Bobot dari unit xi ke unit zj wjk = Bobot dari unit zj ke unit yk
voj = Bobot dari bias ke unit zj
wok= Bobot dari bias ke unit yk
Pengujian
Proses pengujian atau identifikasi merupakan tahap dimana model JST yang telah mengalami pelatihan akan berusaha mengenali pola-pola unik dari data sekuens DNA yang menjadi masukan, dan akan mengklasifikaskan data sekuens DNA tersebut ke dalam masing-masing genus.
Proses identifikasi pada jaringan syaraf tiruan dilakukan melalui proses matematis yang sama dengan operasi arah maju (feed forward), yaitu mengalikan neuron-neuron masukan terhadap bobot jaringan dan ditambah dengan bobot bias untuk masing-masing unit neuron tersembunyi dan keluaran. Nilai bobot diperoleh pada proses pelatihan sebelumnya. Jadi, pada proses identifikasi tidak terjadi perubahan atau penyesuaian bobot.
Proses identifikasi inilah yang dijadikan dasar dalam menentukan data sekuens DNA akan masuk ke dalam kategori yang sesuai dengan genusnya.
Analisis
Langkah pertama dalam tahap analisis adalah menghitung nilai sensitivity
11
sensitivity
1=
tp1 tp
1+fn1
Adapun persamaan dari nilai specificity untuk genus 1 adalah:
specificity1= tn1 tn1+fp1
Pada penelitian ini, yang dimaksud dengan nilai sensitivity1 adalah, perbandingan antara jumlah sekuens DNA uji genus 1 yang terdeteksi sebagai sekuens DNA genus 1 dengan jumlah seluruh sekuens DNA uji genus 1, sedangkan yang dimaksud dengan nilai specificity1 adalah, perbandingan antara jumlah sekuens DNA uji bukan genus 1 yang terdeteksi sebagai bukan sekuens DNA genus 1 dengan jumlah seluruh sekuens DNA uji yang terdeteksi sebagai bukan sekuens DNA genus 1.
Berdasarkan jumlah kelas yang digunakan, pada penelitian ini diperoleh tiga nilai sensitivity dan tiga nilai specificity. Ketiga nilai sensitivity dan specificity
tersebut akan dirata-rata sehingga diperoleh nilai sensitivity dan specificity akhir yang merepresentasikan nilai sensitivity dan specificity penelitian secara keseluruhan. Nilai sensitivity dan specificity digunakan untuk mengetahui seberapa besar kemampuan metode yang digunakan dalam penelitian ini, mampu mengidentifikasi kelas dari sekuens DNA uji, dari seluruh sekuens DNA yang diujikan.
Tabel 5 Confusion matrix genus 1
Keterangan:
tp1 : true positive 1 (jumlah sekuens DNA uji genus 1 yang berhasil teridentifikasi sebagai sekuens DNA genus 1).
tn1 : true negative 1 (jumlah bukan sekuens DNA uji genus 1 yang berhasil teridentifikasi sebagai bukan sekuens DNA genus 1).
fp1 : false positive 1 (jumlah bukan sekuens DNA uji genus 1 yang berhasil teridentifikasi sebagai sekuens DNA genus 1).
fn1 : false negative 1 (jumlah sekuens DNA uji genus 1 yang teridentifikasi sebagai bukan sekuens DNA genus 1).
SekuensDNA uji genus 1
Bukan sekuens DNA uji genus 1 Terdeteksi sebagai sekuens
DNA genus 1 tp1 fp1
Terdeteksi sebagai bukan
12
HASIL DAN PEMBAHASAN
Praproses Data
Data yang digunakan pada penelitian ini untuk known organisms dan new organisms terdiri atas tiga jenis genus, yaitu: Bacillus, Burkholderia, dan
Pseudomonas dengan 4 panjang fragmen yang dipakai yaitu 100 bp, 400 bp, 800
bp, dan 1 Kbp. Setiap sekuens DNA memiliki informasi sources DNA. Pada tahap praproses data, informasi sources DNA akan dipisahkan dari sekuens DNA. Hal ini dikarenakan informasi yang dibutuhkan untuk melakukan proses pelatihan JST dan pengujian data uji hanya kode basa yang ada di setiap sekuens DNA. Hasil dari tahap praproses data adalah sekuens DNA yang telah terpisahkan dari
sources-nya. Ketiga genus data sekuens DNA dengan panjang fragmen yaitu 100
bp, 400 bp, 800 bp, dan 1 Kbp dapat dilihat pada Tabel 6 – 9.
Tabel 6 Tiga genus data sekuens DNA dengan panjang fragmen 100 bp Genus Sekuens DNA untuk known
organisms
Sekuens DNA untuk new organisms
Bacillus 600 sekuens 146 sekuens
Burkholderia 600 sekuens 102 sekuens
Pseudomonas 600 sekuens 112 sekuens
Total 1800 sekuens 360 sekuens
Tabel 7 Tiga genus data sekuens DNA dengan panjang fragmen 400 bp Genus Sekuens DNA untuk known
organisms
Sekuens DNA untuk new organisms
Bacillus 545 sekuens 147 sekuens
Burkholderia 600 sekuens 91 sekuens
Pseudomonas 590 sekuens 109 sekuens
Total 1735 sekuens 347 sekuens
Tabel 8 Tiga genus data sekuens DNA dengan panjang fragmen 800 bp Genus Sekuens DNA untuk known
organisms
Sekuens DNA untuk new organisms
Bacillus 575 sekuens 151 sekuens
Burkholderia 630 sekuens 95 sekuens
Pseudomonas 585 sekuens 112 sekuens
13 Tabel 9 Tiga genus data sekuens DNA dengan panjang fragmen 1 Kbp
Genus Sekuens DNA untuk known
organisms
Sekuens DNA untuk new organisms
Bacillus 540 sekuens 166 sekuens
Burkholderia 650 sekuens 96 sekuens
Pseudomonas 600 sekuens 96 sekuens
Total 1790 sekuens 358 sekuens
Ekstraksi Ciri Spaced K-Mers
Pada penelitian ini, nilai w dan d yang akan digunakan untuk metode
spaced k-mers, yaitu w = 3 dan d = 0, 1, dan 2. Berdasarkan banyaknya basa penyusun sekuens DNA dan nilai w = 3 dan d = 0, 1, dan 2 pada metode spaced k-mers, maksimal banyaknya kombinasi fitur dari sekuens DNA yang dapat dibentuk adalah 192 fitur.
Pada penelitian ini, data yang digunakan terdiri atas tiga jenis genus bakteri yaitu Bacillus, Burkholderia, dan Pseudomonasdengan panjang fragmen 100 bp, 400
bp, 800 bp, dan 1 Kbp. Hasil atau nilai spaced k-mers pada data sekuens DNA
digabung menjadi sebuah matriks berdimensi n × 192. Dimensi baris = n
menunjukan urutan dari data sekuens DNA, sedangkan dimensi kolom = 192 menunjukan urutan kombinasi fitur.
5-Fold Cross Validation
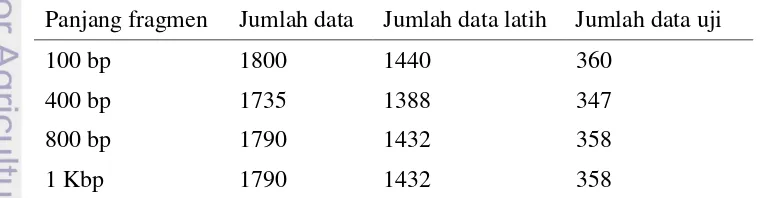
Pada penelitian ini, metode yang digunakan untuk membagi data latih dan data uji adalah k-fold cross validation. Metode tersebut digunakan dengan tujuan agar semua data sekuens DNA pernah menjadi data latih dan data uji, sehingga nilai akurasi yang dihasilkan dapat merepresentasikan nilai akurasi data secara keseluruhan. Pada penelitian ini nilai k yang digunakan adalah 5, sehingga proporsi data pada known organisms untuk data latih adalah 80% dan proporsi data untuk data uji adalah 20%. Jumlah sekuens DNA dari ketiga genus dengan panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp yang akan menjadi data latih dan data uji dapat dilihat pada Tabel 10.
Tabel 10 Jumlah data latih dan data uji dari ketiga genus Panjang fragmen Jumlah data Jumlah data latih Jumlah data uji
100 bp 1800 1440 360
400 bp 1735 1388 347
800 bp 1790 1432 358
14
Klasifikasi JST
Metode klasifikasi yang digunakan pada penelitian ini adalah JST. Terdapat dua tahap dalam melakukan proses klasifikasi, yaitu tahap pelatihan data latih dan pengujian data uji. Proses pelatihan ini ditujukan agar jaringan dapat mempelajari karakteristik setiap genus, berdasarkan data latih yang telah dikelompokkan dengan target yang telah dibuat, sehingga didapatkan suatu jaringan terbaik yang diharapkan mampu mengidentifikasi jenis genus dari suatu data sekuens DNA. Matriks target tersebut dibuat berdasarkan matriks data latih. Matriks target digunakan untuk memberikan informasi kepada jaringan bahwa, suatu kolom pada matriks data latih, termasuk ke dalam genus pertama (Bacillus), genus kedua (Burkholderia), atau genus ketiga (Pseudomonas).
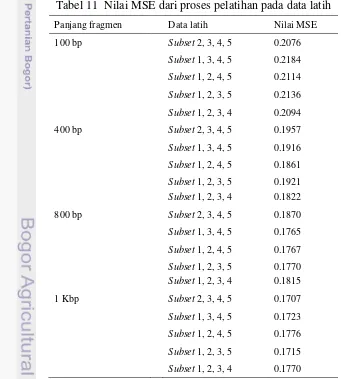
Pada penelitian ini, motode yang digunakan pada tahap pemilihan data latih dan data uji adalah 5-fold cross validation, sehingga terdapat 5 kali proses pelatihan data latih dan pengujian data uji, di setiap panjang fragmen sekuens DNA yang berbeda. Setiap kali proses pelatihan data latih, dihasilkan nilai MSE sebagai indikator baik atau buruknya model jaringan yang dihasilkan. Model jaringan dikatakan baik jika memiliki nilai MSE yang kecil. Nilai MSE terkecil yang diperoleh dari proses pelatihan data latih pada penelitian ini dapat dilihat pada Tabel 11.
Tabel 11 Nilai MSE dari proses pelatihan pada data latih
Panjang fragmen Data latih Nilai MSE
15 Pengujian
Input dari proses pengujian adalah data uji sekuens DNA, berserta jaringan terbaik yang diperoleh pada tahap pelatihan JST. Proses pengujian ini, akan diberlakukan untuk seluruh data uji sekuens DNA pada known organisms dan data uji sekuens DNA pada new organisms dari ketiga genus yang memiliki panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp
Pada proses pengujian, data uji akan melalui tahapan feed forward seperti yang terjadi pada data latih. Namun hanya dilakukan satu kali iterasi. Pada saat iterasi tersebut, input sekuens DNA uji akan dikalikan dengan bobot-bobot yang ada pada jaringan. Bobot-bobot tersebut merupakan bobot yang dihasilkan pada tahapan back propagation pada saat melakukan pelatihan data latih.
Hasil proses pengujian terdiri dari 3 neuron, karena fungsi aktivasi yang digunakan pada penelitian ini adalah fungsi aktivasi sigmoid logaritmik, nilai maksimum yang ada pada ketiga neuron tersebut adalah 1, sedangkan nilai minimum pada ketiga neuron tersebut adalah 0. Pada penelitian ini neuron pertama menunjukan genus Bacillus, neuron kedua menunjukan genus
Burkholderia, dan neuron ketiga menunjukan genus Pseudomonas. Nilai
maksimum yang ada diantara ketiga neuron tersebut menunjukan bahwa sekuens DNA uji masuk ke dalam genus yang nilai neuronnya maksimum tersebut.
Analisis Hasil
Hasil dari proses pengujian selanjutnya akan dihitung dengan menggunakan tabel confusion matrix. Tabel confusion matrix tersebut dibutuhkan untuk melakukan proses perhitungan sensitivity dan specificity. Hasil dari tabel
confusion matrix untuk known organisms dan confusion matrix untuk new organisms dapat dilihat pada Lampiran 5 dan Lampiran 6.
1 Pengujian menggunakan data known organisms
Pengujian menggunakan known organisms akan didapatkan nilai sensitivity
16
Tabel 12 Nilai sensit
setiap genus
Agar perbandingan ni lebih jelas, maka nilai tersebut
sensitivity dan specificity se 800 bp, dan 1 Kbp dapat dil pada Gambar 4, dapat diliha fragmen 100 bp, 400 bp, 800 sedangkan berdasarkan graf tertinggi baik pada panjang terletak pada genus Bacillus
Gambar 4 Grafik nilai berdasarkan pa
nsitivity dan specificity untuk known organisms
nus
Genus Sensitivity Specific Bacillus sebut akan disajikan ke dalam bentuk grafik. G
setiap genus dengan panjang fragmen 100 bp, dilihat pada Gambar 4 dan Gambar 5. Berdasar
ilihat bahwa nilai sensitivity tertinggi baik pad , 800 bp, maupun 1 Kbp, terletak pada genus grafik pada Gambar 5, dapat dilihat bahwa nilai ang fragmen 100 bp, 400 bp, 800 bp, maupun
llus.
Gambar 5 Grafik ni berdasar 2 Pengujian menggu
Pengujian mengg dan specificity. Nila dihasilkan untuk setia dan 1 Kbp dapat diliha
Tabel 13 Nilai se
sensitivity dan specifi
800 bp, dan 1 Kbp dap
k nilai specificity untuk known organisms setiap ge sarkan panjang fragmen
ggunakan data new organisms
enggunakan new organisms akan didapatkan ilai sensitivity dan specificity pada new or
etiap genus dengan panjang fragmen 100 bp, 400 lihat pada Tabel 13.
sensitivity dan specificity untuk new organisms
en Genus Sensitivity Spe
Bacillus
nilai tersebut akan disajikan kedalam bentuk gra
cificity setiap genus dengan panjang fragmen 100 bp dapat dilihat pada Gambar 6 dan Gambar 7. Ber
100 bp 400 bp 800 bp 1 Kbp
18
pada Gambar 6, dapat diliha fragmen 100 bp, 400 bp, 800 sedangkan berdasarkan graf tertinggi baik pada panjang
Bacillus, dan pada panjang
Burkholderia.
Gambar 6 Grafik ni setiap ge
Gambar 7 Grafik ni setiap ge 3 Nilai rata-rata sensitivit
Untuk menentukan pa dibutuhkan nilai rata-rata da dan specificity dari ketiga je berdasarkan panjang fragm Tabel 15.
ilihat bahwa nilai sensitivity tertinggi baik pad , 800 bp, maupun 1 Kbp, terletak pada genus grafik pada Gambar 7, dapat dilihat bahwa nilai jang fragmen 100 bp dan 1 Kbp terletak pa
ang fragmen 400 bp dan 800 bp terletak pa
ik nilai sensitivity untuk new organisms
p genus berdasarkan panjang fragmen
ik nilai specificity untuk new organisms
p genus berdasarkan panjang fragmen
ivity dan specificity
n panjang fragmen terbaik secara umum bagi ket dari sensitivity dan specificity. Nilai rata-rata ga jenis genus untuk known organisms dan new
Tabel
sensitivity dan specif
dilihat pada Gambar 8 d
Gambar 8
bel 14 Nilai rata-rata sensitivity dan specificity
untuk known organisms dari ketiga jenis genus
njang fragmen Sensitivity Specificity
100 bp 0.8306 0.9153
400 bp 0.9248 0.9617
800 bp 0.9564 0.9780
Kbp 0.9716 0.9854
bel 15 Nilai rata-rata sensitivity dan specificity
untuk new organisms dari ketiga jenis genus
anjang fragmen Sensitivity Specificity
100 bp 0.7622 0.8959
400 bp 0.8369 0.9351
800 bp 0.9039 0.9615
1 Kbp 0.9211 0.9690
ndingan nilai rata-rata sensitivity dan specificit organisms dari ketiga jenis genus terlihat le n disajikan kedalam bentuk grafik. Grafik
cificity berdasarkan panjang fragmen yang di r 8 dan Gambar 9.
Grafik nilai rata-rata sensitivity dari ketiga ge
20
Gambar 9 Grafi Berdasarkan grafik pa panjang fragmen terbaik
Bacillus, Burkholderia, dan nilai sensitivity untuk know
0.9211, sedangkan nilai spe
adalah 0.9854 dan 0.9690 menghasilkan fitur ekstr merepresentasikan karakte (Bacillus, Burkholderia, dan
Dapat kita lihat jug bertambahnya panjang frag semakin banyak jumlah pa genetik yang lebih banyak JST, tidak sulit untuk me
afik nilai rata-rata specificity dari ketiga genus k pada Gambar 8 dan Gambar 9 dapat diliha
k untuk mengidentifikasi sekuens DNA pa dan Pseudomonas adalah panjang fragmen 1 Kbp,
nown organisms dan new organisms adalah 0.9
specificity untuk known organisms dan new
690. Hal ini terjadi karena, panjang fragme straksi dengan informasi terlengkap yang kteristik yang berbeda bagi ketiga genus , dan Pseudomonas).
uga bahwa grafik tersebut cenderung mena ragmen dari sekuens DNA. Hal ini menandaka panjang fragmen, maka dapat memberikan ak pada suatu organisme. Sehingga pada tahap mencari perbedaan atau mempelajari karakter
SIMPULAN DAN SARAN
Simpulan
21 2 Nilai sensitivity dan specificity terbaik yang mampu dicapai oleh metode
ekstraksi ciri spaced k-mers untuk new organisms diperoleh pada panjang fragmen 1 Kbp, yaitu dengan nilai sensitivity 0.9211 dan nilai specificity
0.9690.
Saran
Ada beberapa hal yang dapat dilakukan untuk melanjutkan topik penelitian ini, yaitu:
1 Menambah jumlah data dengan jenis genus bakteri yang lebih beragam.
2 Melakukan klasifikasi hingga batasan tingkat taksonomi yang lebih spesifik, seperti klasifikasi dari tingkat genus hingga tingkat spesies.
3 Melakukan klasifikasi multi organisme, yaitu klasifikasi di antara beberapa jenis organisme yang berbeda, seperti klasifikasi antara sekuens DNA bakteri dengan sekuens DNA virus.
DAFTAR PUSTAKA
Kusuma, WA. 2012. Combined approaches for improving the performance of de novo DNA sequence assembly and metagenomic classification of short fragments from next generation sequencer [disertasi]. Tokyo (JP): Tokyo Institute of Technology.
McHardy AC, Martin HG, Tsirigos A, Hugenholtz P, Rigoutsos I. 2007. Accurate phylogonetic classification of variabel-length DNA fragments. Nature Methods. 4(1):63-72. doi:10.1038/nmeth976.
Osadebey ME. 2006. Integrated content-based image retrieval using texture, shape and spatial information [tesis]. Umeå (SE): Umeå University.
Richter DC, Ott F, Auch AF, Schmid R, Huson DH. 2009. User manual for MetaSim V0.9.5 [Internet]. [diunduh 2012 Nov 27]. Tersedia pada: http://www-ab.informatik.uni.tuebigen.de/software/metasim.
Riesenfeld CS, Schloss PD, Handelsman J. 2004. Metagenomics: genomic analysis of microbial communities. Annual Review Genetics. 38:525-553. Wooley JC, Godzik A, Friendberg I. 2010. A primer on metagenomics. PLos
Computational Biology. 6(2):1-13. doi:10.1371/journal.pcbi.1000667.
Yani E. 2005. Pengantar jaringan syaraf tiruan [Internet]. [diunduh 2013 Feb 11]. Tersedia pada:
22
Lampiran 1 Daftar organisme untuk known organisms untuk setiap genus dengan panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp
Nama organisme pada genus Bacillus Bacillus amyloliquefaciens FZB42' Bacillus anthracis str. Ames Ancestor' Bacillus anthracis str. Ames chromosome' Bacillus anthracis str. Sterne chromosome' Bacillus cereus ATCC 10987 chromosome' Bacillus cereus ATCC 14579'
Bacillus cereus E33L'
Bacillus cereus subsp. cytotoxis NVH 391-98' Bacillus clausii KSM-K16'
Bacillus halodurans C-125 chromosome' Bacillus licheniformis ATCC 14580'
Bacillus subtilis subsp. subtilis str. 168 chromosome'
Bacillus thuringiensis serovar konkukian str. 97-27 chromosome' Bacillus thuringiensis str. Al Hakam chromosome'
Bacillus weihenstephanensis KBAB4'
Nama organisme pada genus Burkholderia
Burkholderia ambifaria AMMD chromosome chromosome 1' Burkholderia ambifaria MC40-6 chromosome chromosome 1' Burkholderia cenocepacia AU 1054 chromosome 3'
Burkholderia cenocepacia HI2424 chromosome chromosome 1' Burkholderia cenocepacia J2315 chromosome chromosome 1' Burkholderia cenocepacia MC0-3 chromosome chromosome 1' Burkholderia mallei ATCC 23344 chromosome chromosome 1' Burkholderia mallei NCTC 10229 chromosome I'
Burkholderia mallei NCTC 10247 chromosome I' Burkholderia mallei SAVP1 chromosome I'
Burkholderia multivorans ATCC 17616 chromosome chromosome 1'
Burkholderia phymatum STM815 chromosome chromosome 1' Burkholderia phytofirmans PsJN chromosome chromosome 1' Burkholderia pseudomallei 1106a chromosome I'
Burkholderia pseudomallei 1710b chromosome chromosome I' Burkholderia pseudomallei 668 chromosome I'
Burkholderia pseudomallei K96243 chromosome chromosome 1' Burkholderia sp. 383 chromosome 1'
Burkholderia sp. 383 chromosome chromosome 2'
23 Lampiran 1 Lanjutan
Nama organisme pada genus Pseudomonas Pseudomonas aeruginosa PA7'
Pseudomonas aeruginosa PAO1 chromosome' Pseudomonas aeruginosa UCBPP-PA14' Pseudomonas fluorescens Pf-5 chromosome' Pseudomonas fluorescens Pf0-1 chromosome' Pseudomonas putida F1 chromosome'
Pseudomonas putida GB-1 chromosome' Pseudomonas putida KT2440 chromosome' Pseudomonas putida W619 chromosome'
Pseudomonas syringae pv. phaseolicola 1448A chromosome' Pseudomonas syringae pv. syringae B728a'
Pseudomonas syringae pv. tomato str. DC3000 chromosome'
24
Lampiran 2 Daftar organisme untuk new organisms untuk setiap genus dengan panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp
Nama organisme pada genus Bacillus Bacillus amyloliquefaciens DSM 7' Bacillus anthracis str. A0248' Bacillus anthracis str. CDC 684' Bacillus atrophaeus 1942 chromosome'
Bacillus cellulosilyticus DSM 2522 chromosome' Bacillus cereus 03BB102'
Bacillus cereus AH187 chromosome' Bacillus cereus AH820'
Bacillus cereus B4264' Bacillus cereus G9842'
Bacillus cereus Q1 chromosome'
Nama organisme pada genus Burkholderia Burkholderia cepacia AMMD chromosome 2'
Burkholderia glumae BGR1 chromosome chromosome 1' Burkholderia rhizoxinica HKI 454 chromosome'
Burkholderia rhizoxinica HKI 454 plasmid pBRH01' Burkholderia rhizoxinica HKI 454 plasmid pBRH02' Burkholderia sp. CCGE1001 chromosome 1'
Burkholderia sp. CCGE1001 chromosome chromosome 2' Burkholderia sp. CCGE1002 chromosome 1'
Burkholderia sp. CCGE1002 chromosome 2'
Burkholderia sp. CCGE1003 chromosome chromosome 1' Burkholderia sp. CCGE1003 chromosome chromosome 2' Burkholderia sp. JV3 chromosome'
Nama organisme pada genus Pseudomonas Pseudomonas fulva 12-X chromosome'
25 Lampiran 3 Algoritme pelatihan JST back propagation
Secara rinci algoritme pelatihan jaringan back propagation dapat diuraikan sebagai berikut:
• Langkah 0: Inisialisasi nilai bobot (biasanya digunakan nilai acak yang kecil) • Langkah 1: Selama kondisi berhenti belum dicapai, maka lakukan langkah
ke-2 hingga langkah ke-9.
• Langkah 2: Untuk setiap pasangan pola pelatihan, lakukan langkah ke-3 sampai langkah ke-8.
Tahap I Feed-forward
• Langkah 3: Setiap unit input (xi, i=1,…,n), menerima sinyal input xi dan mengirimkan sinyal tersebut keseluruh unit yang ada pada lapisan tersembunyi.
• Langkah 4: Untuk setiap unit tersembunyi (zj, j=1,…,p), dihitung nilai input
dengan menggunakan nilai bobotnya:
z_inj=v0j+ xi
n
i=1
vij
Kemudian dihitung nilai output dengan menggunakan fungsi aktivasi yang dipilih:
zj = f ( z_inj ).
• Hasil dari fungsi aktivasi tersebut dikirim ke semua unit pada lapis output.
y_in
k = w0k+ zj p
j=1
wjk
Kemudian dihitung nilai output dengan menggunakan fungsi aktivasi:
yk = f y_in
k
Tahap II Back-propagation
• Langkah 6: Untuk setiap unit output (yk, k=1,..,m) menerima pola target yang bersesuaian dengan pola input, dan kemudian dihitung informasi kesalahan:
δk = tk-yk '(y_ink)
Kemudian dihitung koreksi nilai bobot yang kemudian akan digunakan untuk memperbaharui nilai bobot wjk:
∆wjk = αδkzj
Hitung koreksi nilai bias yang kemudian akan digunakan untuk memperbaharui nilai wok:
∆w0k = αδk
26
Lampiran 3 Lanjutan
• Langkah 7: Untuk setiap unit tersembunyi (zj, j=1,…,p) dihitung delta input
yang berasal dari unit pada lapisan output:
δ_in
j = δk m
k=1
wjk
Kemudian nilai tersebut dikalikan dengan nilai turunan dari fungsi aktivasi untuk menghitung informasi kesalahan:
δj = δ_injf(z_inj)
Hitung koreksi nilai bobot yang kemudian digunakan untuk memperbaharui nilai vij:
∆vij =αδjxi
Tahap III Memperbaharui nilai bobot dan bias
• Langkah 8: Tiap nilai bias dan bobot (j=0,…,p) pada unit output (yk,
k=1,…,m) diperbaharui:
wjk new =wjk old +∆wjk vij new =vij old +∆vij
• Langkah 9: Menguji apakah kondisi berhenti sudah terpenuhi.
27 Lampiran 4 Algoritme inisialisasi Nguyen-Widrow
• Tetapkan:
n = jumlah neuron pada input layer p = jumlah neuron pada hidden layer
β = faktor kesalahan (0.7(p)1/n)
• Kerjakan untuk setiap unit pada lapaisan tersembunyi (j = 1,2,…,p):
a. Inisialisasi bobot-bobot dari lapisan input ke lapisan tersembunyi: vij =
bilangan acak (-0.5 – 0.5)
b. Hitung Vj = V21j+V22j+…+V2nj
c. Inisialisasi ulang bobot-bobot: Vij = βVij ||Vij||
28
Lampiran 5 Confusion matrix untuk know organisms darisetiap genus dengan panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp
• Panjang fragmen 100 bp
Subset 1
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 114 5 12
Burkholderia 2 93 16
Pseudomonas 4 22 92
Subset 2
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 110 4 10
Burkholderia 2 105 22
Pseudomonas 8 11 88
Subset 3
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 108 4 7
Burkholderia 4 100 25
Pseudomonas 8 16 88
Subset 4
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 114 6 10
Burkholderia 2 101 24
Pseudomonas 4 13 86
Subset 5
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 112 2 12
Burkholderia 1 90 14
29 Lampiran 5 Lanjutan
• Panjang fragmen 400 bp
Subset 1
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 100 1 4
Burkholderia 3 111 18
Pseudomonas 6 8 96
Subset 2
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 102 1 5
Burkholderia 3 109 3
Pseudomonas 4 10 110
Subset 3
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 107 0 4
Burkholderia 1 111 3
Pseudomonas 1 9 111
Subset 4
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 102 1 4
Burkholderia 5 114 6
Pseudomonas 2 5 108
Subset 5
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 100 2 2
Burkholderia 0 110 3
30
Lampiran 5 Lanjutan
• Panjang fragmen 800 bp
Subset 1
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 113 1 3
Burkholderia 1 122 5
Pseudomonas 1 3 109
Subset 2
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 113 1 2
Burkholderia 0 116 1
Pseudomonas 2 9 114
Subset 3
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 113 0 5
Burkholderia 1 119 3
Pseudomonas 1 7 109
Subset 4
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 112 2 1
Burkholderia 1 120 4
Pseudomonas 2 4 112
Subset 5
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 114 3 6
Burkholderia 0 116 2
31 Lampiran 5 Lanjutan
• Panjang fragmen 1 Kbp
Subset 1
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 107 1 1
Burkholderia 0 127 1
Pseudomonas 1 2 118
Subset 2
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 105 0 2
Burkholderia 2 129 2
Pseudomonas 1 1 116
Subset 3
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 106 2 0
Burkholderia 0 124 6
Pseudomonas 2 4 114
Subset 4
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 105 1 3
Burkholderia 0 124 1
Pseudomonas 3 5 116
Subset 5
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 107 1 0
Burkholderia 0 121 1
32
Lampiran 6 Confusion matrix untuk new organisms darisetiap genus dengan panjang fragmen 100 bp, 400 bp, 800 bp, dan 1 Kbp
• Panjang fragmen 100 bp
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 143 8 6
Burkholderia 1 55 20
Pseudomonas 2 39 86
• Panjang fragmen 400 bp
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 144 0 2
Burkholderia 0 50 0
Pseudomonas 3 41 107
• Panjang fragmen 800 bp
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 151 1 2
Burkholderia 0 71 2
Pseudomonas 0 23 108
• Panjang fragmen 1 Kbp
Genus hasil prediksi
Genus asal Bacillus Burkholderia Pseudomonas
Bacillus 163 1 0
Burkholderia 0 77 2
33