www.elsevier.com / locate / bres
Research report
N-Methyl-
D
-aspartate receptor activation results in regulation of
extracellular signal-regulated kinases by protein kinases and
phosphatases in glutamate-induced neuronal apototic-like death
a,b a a ,
*
bQian Jiang
, Zhenglin Gu , Guangyi Zhang
, Guozhang Jing
a
Research Center for Biochemistry and Molecular Biology, Xuzhou Medical College, 84 West Huai-hai Road, Xuzhou, Jiangsu 221002, PR China
b
Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 294 Tai-yuan Road, Shanghai 200031, PR China
Accepted 19 September 2000
Abstract
Extracellular signal-regulated kinases (ERK1 / ERK2) have been shown transiently activated and involved in excitotoxicity. We searched for upstream molecules responsible for the regulation of glutamate-induced ERK1 / ERK2 activation and ERK1 / ERK2-mediated apototic-like death in cultured rat cortical neurons. ERK1 / ERK2 activation (monitored by anti-active ERK1 / ERK2 antibody) was almost
21
completely prevented by blockage of NMDA receptor (NMDA-R) or elimination of extracellular Ca , but not any other glutamate 21
receptor or L-type voltage-gated Ca channel. It was prevented largely by inhibition of protein kinase C (PKC), protein-tyrosine kinases (PTK), respectively, but mildly by that of CaM kinase II. Combined inhibition of CaM kinase II (but not PTK) and PKC had an additive effect. Reversion of ERK1 / ERK2 activation was largely prevented by inhibition of protein phosphatase (PP) 1 or protein tyrosine phosphatase (PTP). Combined inhibition of PP 1 and PTP had no additive effect. Glutamate-induced apoptotic-like death (determined by DAPI staining) was largely prevented by inhibition of NMDA-R, PKC, CaM kinase II, PTK and MEK1 / MEK2 (ERK1 / ERK2 kinase), respectively. Combined inhibition of CaM kinase II (but not PKC or PTK) and MEK1 / MEK2 had an additive effect. Glutamate-induced apoptotic-like death was promoted by inhibition of PP1 and PTP, respectively. The above results suggested that in glutamate-induced cortical neurotoxicity ERK1 / ERK2 activation be mainly mediated by NMDA-R. Subsequently, a pathway dependent on both PKC and PTK was mainly involved, which was also mainly responsible for ERK1 / ERK2-mediated apoptotic-like death, and a CaM kinase II-dependent pathway was relatively mildly involved. Reversion of ERK1 / ERK2 activation was mainly mediated by a pathway dependent on both PP1 and PTP, which might be involved in the restrain of glutamate-induced neurotoxicity. 2000 Elsevier Science B.V. All rights reserved.
Theme: Neurotransmitters, modulators, transporters, and receptors
Topic: Excitatory amino acids: excitotoxicity
Keywords: Extracellular signal-regulated kinase; Excitotoxicity; Glutamate receptor; Protein kinase; Protein phosphatase; Cortical neuron
1. Introduction intracellular cascades leading to excitotoxicity are largely
21
unknown, potential Ca targets include nitric oxide Glutamate-induced excitotoxicity, with certain charac- synthase (NOS) [2], protein phosphatases and protein teristics of apoptosis [5,25,48], has been implicated in kinases [9,28]. Recently, several molecules including NOS many neuronal degenerative diseases and considered pre- [49,50], protein kinase C (PKC), CaM kinase II [18] and
21
dominantly mediated by an overload of intracellular Ca , some members of protein tyrosine kinases (PTK), such as mainly as a result of influx via N-methyl-D-aspartate PYK2 (proline-rich tyrosine kinase 2) and c-Src
21 21
receptor (NMDA-R) [30,51]. Although the Ca -activated [16,29,39], have been shown to transduce Ca signaling to ERK1 / ERK2 (extracellular signal-regulated kinases) cascade.
*Corresponding author. Tel.:186-516-574-8423; fax: 1
86-516-574-ERK1 / ERK2, with molecular masses of 44 and 42 kDa,
8429.
E-mail address: [email protected] (G. Zhang). respectively, are classical members of mitogen-activated
protein kinase (MAPK) superfamily. Both require specific glutamine, Gibco-BRL). Cultures were used after 13 days diphosphorylation of both threonine and tyrosine residues in vitro when the cells were vulnerable to glutamate insult at the regulatory sites by MEK1 / MEK2 (ERK1 / ERK2 and verified .95% neurofilaments positive by immuno-kinase) for activation [33,42]. ERK1 / ERK2 cascades play staining.
important roles in signal transduction from cell surface to
nucleus. The well-documented neurotropic growth factor 2.2. Drug treatment receptor-mediated activation cascade (Ras / Raf / MEK /
ERK) has been thought to play important roles in cell Excitotoxicity was induced by 50 mM glutamate expo-growth, proliferation and survival [10,18,20,22,32]. sure for 15 min, during which the medium was changed
21
Recently, ERK1 / ERK2 have been found activated after into modified EBSS (Mg -free Earle’s balanced salt relatively mild stimulation of glutamate receptors and solution (EBSS) (Gibco-BRL) supplemented with 5 mM involved in some activity-dependent functions [18,20]. glycine). For restoration, at the end of glutamate exposure, Furthermore, ERK1 / ERK2 have also been found activated cultures were rinsed twice with EBSS, and the original in some excitotoxicity-associated events, such as stroke, feeding medium was restored. For drug treatments, seizure and Alzheimer’s disease [3,19,24]. We previously PD98059 (Calbiochem, San Diego, CA, USA), MK-801 observed that ERK1 / ERK2 were transiently activated in (dizocilpine maleate, RBI, Natick, MA, USA.), L-AP-3 glutamate-induced apoptotic-like death in cultured rat (L-(1)-2-amino-3-phosphono-propionic acid, RBI), cortical neurons, and PD98059, a specific inhibitor for DNQX (6,7,-dinitroquinoxaline-2,3 (1H,4H)-dione, MEK1 / MEK2, completely inhibited such activation and Sigma), nifedipine (Sigma), EGTA (Sigma), genistein partially prevented the glutamate-induced apoptotic-like (Sigma), H-89 (Calbiochem), KN-62 (RBI), sphingosine
v
death [26]. Therefore, ERK1 / ERK2 might be excessively (RBI),L-NNA (N -nitro-L-argine, Sigma) were added form activated transiently and involved in the glutamate-induced 20 min before till the end of glutamate exposure,
respec-cortical neurotoxicity. tively, or in combination. In some other cases, sodium
However, little is known about the upstream cascade of orthovanadate (Sigma), okadaic acid (RBI) and cyclos-the variation of ERK1 / ERK2 in excitotoxicity. In this porin A (Sigma) were added from 20 min before until 3 h study, we searched for upstream molecules responsible for after glutamate exposure, respectively. PD98059, DNQX, the variation of ERK1 / ERK2 and the ERK1 / ERK2-me- nifedipine, genistein, sphingosine, okadaic acid and cyclos-diated apoptotic-like death in the glutamate-induced neuro- porin A were made as 5003 stocks in dimethyl sulfoxide toxicity in cultured rat cortical neurons. Several molecules (DMSO), respectively. KN-62 was made as 3003 stocks were investigated, including three subtypes of glutamate in methanol. Other drugs were made as 2003 stocks in
21
receptors, L-type voltage-gated Ca channel (L-VGCC), water. Vehicle controls were treated only with 50 mM NOS, some protein kinases and protein phosphatases, each glutamate and vehicle (0.5% water or 0.2% DMSO or of which has been implicated involved either in excitotox- 0.3% methanol) in modified EBSS. Sham controls were icity or in the regulation of ERK1 / ERK2 activation in treated only with modified EBSS.
other cases.
2.3. Cell extracts preparation and Western immunoblot
2. Materials and methods Cultured cells were rinsed with PBS, scraped off the
wells. Each sample was pooled from two wells
(approxi-7
2.1. Neuronal cultures mately 1.2310 cells) and homogenized in 160ml ice-cold
buffer (50 mM 3-(N-morpholino) propane-sulfonic acid, Cortical neuronal cultures were prepared from 17-day- MOPS, pH 7.4), 0.5 mM dithiothreitol, 2 mM sodium old Sprague–Dawley rat embryos as previously described orthovanadate, 0.5 mM EDTA, 1 mM EGTA, 0.5 mM [6]. Briefly, neocortex was meticulously isolated in ice- ouabain, 1 mM phenylmethylsulfonyl fluoride, 0.5 mM cold high glucose Dulbecco’s modified Eagle medium leupeptin and 0.5 mM pepstatin A, and centrifuged at (h-DMEM, Gibco-BRL, Grand Island, NY, USA). Cortical 15 0003g for 15 min at 48C. Ten ml were removed for cells were dissociated by trypsinization (0.25% (w / v) in protein concentration determination by Lowry method
21 21
Ca - and Mg -free Hank’s balanced salt solution (Gib- [31], the remaining supernatant was incubated in sample co-BRL), at 378C for 15 min), followed by gentle triturat- buffer (2% sodium dodecyl sulfate, 20% glycerol, 5% ing in plating medium (h-DMEM supplemented with 10% b-mercaptoethanol, 62.5 mM Tris–HCl, pH 6.8, and fetal bovine serum and 10% horse serum, Gibco-BRL). 0.01% bromphenol blue) at 968C for 5 min. Equal amount Cells were seeded onto poly-L-lysine (Sigma, St. Louis, of proteins (40mg) were separated by 10% SDS–PAGE by MO, USA)-coated wells or coverslips at a density of the method of Laemmli [27] and electrotransferred onto
5 2
polyclonal, 1:10 000) or anti-active (diphosphorylated) ERK1 / ERK2 antibody (Sigma, monoclonal, 1:5000) at 48C overnight. Detection was carried out by alkaline phosphatase-conjugated goat anti-rabbit IgG (Sigma, 1:20 000) or goat anti-mouse IgG (Sigma, 1: 40 000) and developed with NBT / BCIP color substrate (Sigma). After immunoblot, bands on filter were scanned, quantitative analyzed and printed by an image analyzer affiliated with digital graphic printer (LabWorks Software, UVP, Upland, CA, USA). Protein level and activation (diphosphorylation) level of ERK1 / ERK2, based on immunoreactivities of ERK1 / ERK2 and active ERK1 / ERK2, respectively, were expressed as fold versus sham control of optical density of certain band from Western immunoblot.
2.4. Assessment of apoptotic-like cell death
5
Live cells grown on each coverslip (2310 cells) were incubated with 10 mg / ml fluorescent DNA binding dye DAPI (49,6-diamidino-2-phenylindole, Sigma) at 378C for 30 min, washed with PBS and excited with vertical fluorescent at 400 nm. With fluorescence collected at 455 nm, apoptotic-like cells were characterized by the presence
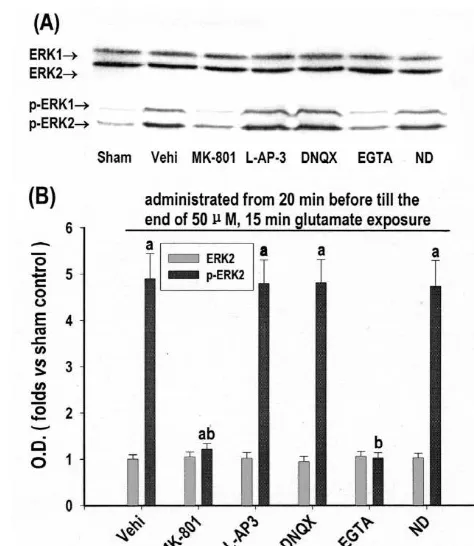
of condensed and fragmented nuclei, as opposed to the Fig. 1. Effects of antagonists of glutamate receptors, L-VGCC and
21
extracellular Ca elimination on ERK1 / ERK2 activation in
glutamate-diffuse staining observed in nonapoptotic cells. Each
induced apoptotic-like death in cultured rat cortical neurons. Thirteen
sample was pooled from three coverslips. The proportion
days in vitro cortical neurons were exposed to 50mM glutamate for 15
of apoptotic-like cells was calculated as a percentage of
min. MK-801 (20mM), L-AP-3 (1 mM), DNQX (20mM), nifedipine
total cells counted in 10 microscopic fields (3400). (ND, 20mM) and EGTA (5 mM) were added, respectively, 20 min
before and during the glutamate exposure. (A) Western immunoblot at 15 min of glutamate exposure with anti-ERK1 / ERK2 antibody (top) or
2.5. Statistics
anti-diphosphorylated ERK1 / ERK2 (p-ERK1 / ERK2) antibody (bottom). (B) Quantitative representations expressed as fold versus sham control
Values were expressed as mean6S.D. from five
in-(Sham, treated with no drug) of optical density (O.D.) ERK2 or of
dependent cultures. One-way ANOVA was used. Com- p-ERK2 band from Western immunoblot. Each point represents
a b
parisons of each group to control were by LSD (least mean6S.D. of five independent cultures. P,0.05 versus sham, P,0.05 versus vehicle control (Vehi, treated only with glutamate and vehicle).
significant difference) test. Others were by q test (New-man–Keuls test). A P value of ,0.05 was considered significant.
(1 mM) [41], or KA /AMPA-R with DNQX (20mM) [23], or L-VGCC with nifedipine (20 mM) [44]. Since the
3. Results alterations of p-ERK1 and p-ERK2 were much the same,
we directed attention to p-ERK2, which displayed a 3.1. The role of glutamate receptors,L-VGCC and relatively stronger immunoreactivity.
21
extracellular Ca in ERK1 /ERK2 activation in
glutamate-induced apoptotic-like death in cultured rat 3.2. The role of protein kinases and NOS in ERK1 /
cortical neurons ERK2 activation in glutamate-induced apoptotic-like
death in cultured rat cortical neurons
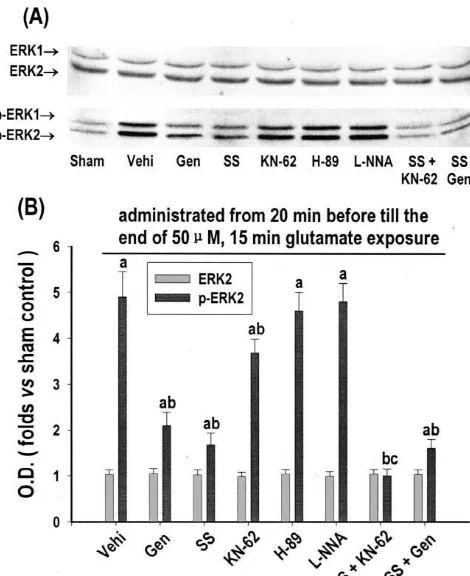
Protein level of ERK1 / ERK2, as indicated by ERK1 /
ERK2 immunoreactivities, was unaffected (Fig. 1A, top, As shown in Fig. 2, ERK1 / ERK2 activation was largely and B). While activation level of ERK1 / ERK2, as indi- prevented by inhibition of either PTK with genistein (100 cated by diphosphorylated ERK1 / ERK2 (p-ERK1 / ERK2), mM, also as an inhibitor of topoisomerase II kinase) [1] or was increased to 4.9-fold versus sham control at 15 min of PKC with sphingosine (10 mM) [12]. It was weakly but glutamate exposure (Fig. 1A, bottom, and B). Such still significantly prevented by inhibition of CaM kinase II activation was almost totally prevented by either blockage with KN-62 (40mM) [45], but not significantly affected by of NMDA-R with MK-801 (20 mM) or elimination of inhibition of PKA with H-89 (40mM, also as an inhibitor
21
21
Fig. 3. Effects of NMDA-R antagonist, extracellular Ca eliminator, inhibitors of NOS and protein kinases on glutamate-induced apoptotic-like death in cultured rat cortical neurons. Thirteen days in vitro cortical neurons were exposed to 50mM glutamate for 15 min. MK-801 (20mM), EGTA (5 mM),L-NNA (20mM), genistein (Gen, 100mM), KN-62 (40
mM), sphingosine (SS, 10mM) and PD98059 (PD, 50mM) were added into the medium 20 min before and during the glutamate exposure, respectively. DAPI staining at 18 h after exposure were quantitative represented as percentage of total cells counted in 10 microscopic fields (3400). Each point represents mean6S.D. of five independent cultures.
a b
P,0.05 versus sham control (Sham, with no drug treatment), P,0.05 Fig. 2. Effects of inhibitors of protein kinase and NOS on ERK1 / ERK2
versus vehicle control (Vehi, treated only with glutamate and vehicle), activation in glutamate-induced apoptotic-like death in cultured rat c
P,0.05 versus single drug treatment. cortical neurons. Thirteen days in vitro cortical neurons were exposed to
50mM glutamate for 15 min. Genistein (Gen, 100mM), H-89 (40mM), KN-62 (40mM), sphingosine (SS, 10 mM) andL-NNA (20 mM) were
added, respectively, 20 min before and during the glutamate exposure. (A) PD98059 (50mM) [26], or NOS withL-NNA (100 mM).
Western immunoblot at 15 min of glutamate exposure with anti-ERK1 / A significant additive effect was observed in combined ERK2 antibody (top) or anti-diphosphorylated ERK1 / ERK2 (p-ERK1 /
inhibition of CaM kinase II (but not PKC or PTK) and
ERK2) antibody (bottom). (B) Quantitative representations expressed as
ERK1 / ERK2. The number of total cells counted in 10
fold versus sham control (Sham, treated with no drug) of optical density
microscopic fields (3400) is about 34006360 cells.
(O.D.) of ERK2 or p-ERK2 band from Western immunoblot. Each point
a
represents mean6S.D. of five independent cultures. P,0.05 versus sham,
b
P,0.05 versus vehicle control (Vehi, treated only with glutamate and
c
vehicle), P,0.05 versus single drug treatment. 3.4. The role of protein phosphatases in the reversion of
ERK1 /ERK2 activation in glutamate-induced apoptotic-that of either sphingosine or genistein. Combined inhibi- like death in cultured rat cortical neurons
tion of CaM kinase II and PKC had a significantly additive
and completely preventive effect. Combined inhibition of As shown in Fig. 4, at 3 h after glutamate exposure, PTK and PKC had no significant additive effect. ERK1 / ERK2 activation reverted to sham control level. Such reversion was largely prevented by inhibition of both
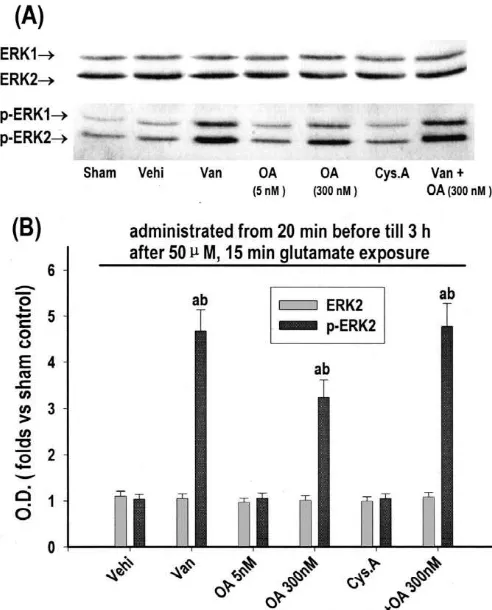
21
3.3. The role of NMDA-R, extracellular Ca , PKC, protein phosphatase (PP) 1 and PP2A with 300 nM
PTK, CaM kinase II and NOS in ERK1 /ERK2-mediated okadaic acid [4], or protein tyrosine phosphatase (PTP)
apoptotic-like death in glutamate-induced excitotoxicity with sodium orthovanadate (200 mM, also as an inhibitor
in cultured rat cortical neurons of ATPase and alkaline phosphatase) [15], but not
sig-nificantly affected by PP2A with 5 nM okadaic acid [4], or As determined by DAPI staining (Fig. 3), at 18 h after PP 2B with cyclosporin A (2 mM, also as an immuno-glutamate exposure, the number of apoptotic-like cells was suppressant) [17]. Combined use of okadaic acid (300 nM) significantly increased to 82% versus sham control (11%). and sodium orthovanadate (200mM) had no significantly Such increase was significantly prevented by elimination additive effect. The concentration of each drug shown in
21
Fig. 5. Effects of inhibitors of protein phosphatases on glutamate-induced apoptotic-like death in cultured rat cortical neurons. Thirteen days in vitro cortical neurons were exposed to 50mM glutamate for 15 min. Okadaic acid (OA, 300 nM) and sodium orthovanadate (Van, 200mM) were added from 20 min before until 3 h after glutamate exposure, respectively. DAPI staining at 18 h after exposure were quantitative represented as per-centage of total cells counted in 10 microscopic fields (3400). Each point
a
represents mean6S.D. of five independent cultures. P,0.05 versus sham
b
control (Sham, with no drug treatment), P,0.05 versus vehicle control
c
(Vehi, treated only with glutamate and vehicle), P,0.05 versus single drug treatment.
Fig. 4. Effects of inhibitors of protein phosphatases on the reversion of death in cultured rat cortical neurons, and such activation ERK1 / ERK2 activation in glutamate-induced apoptotic-like death in
was almost completely prevented by either blockage of
cultured rat cortical neurons. Thirteen days in vitro cortical neurons were 21
NMDA-R or elimination of extracellular Ca [26]. L
-exposed to 50mM glutamate for 15 min. Sodium orthovanadate (Van,
VGCC and glutamate receptors, including NMDA-R, a
-200 mM), okadaic acid (OA, 5 nM), okadaic acid (OA, 300 nM) and
cyclosporin A (Cys A, 2 mM) were added, respectively, from 20 min amino-3-hydroxy-5-methyl-4-isoxazolepropionate / kainate
before until 3 h after the glutamate exposure. (A) Western immunoblot at receptor (AMPA / KA-R) and metabotropic glutamate re-3 h after glutamate exposure with anti-diphosphorylated anti-ERK1 /
ceptor (mGlu-R), have all been shown involved in ERK1 /
ERK2 antibody (top) or ERK1 / ERK2 (p-ERK1 / ERK2) antibody
(bot-ERK2 activation in some other cases [18,20]. However, in
tom). (B) Quantitative representations expressed as fold versus sham
this study, blockage of none of them, except for NMDA-R,
control (Sham, treated with no drug) of optical density (O.D.) of ERK2 or
p-ERK2 band from Western immunoblot. Each point represents significantly affected ERK1 / ERK2 activation. Therefore,
a b
mean6S.D. of five independent cultures. P,0.05 versus sham, P,0.05 in glutamate-induced cortical neurotoxicity ERK1 / ERK2 versus vehicle control (Vehi, treated only with glutamate and vehicle).
activation might be mainly mediated by NMDA-R-induced
21
influx of extracellular Ca .
3.5. The role of PP1 and PTP in glutamate-induced Stimulation of NMDA-R has been shown to mediate
excitotoxicity in cultured rat cortical neurons ERK1 / ERK2 activation, which might involve PKC and CaM kinase II in hippocampal neurons [18] and PTK in As shown in Fig. 5, inhibition of PP1 with 300 nM striatal neurons [47]. However, the upstream cascade(s) is okadaic acid and PTP with sodium orthovanadate (200 unknown in excitotoxicity. In this study, in
glutamate-mM) can promote glutamate-induced apoptotic-like death induced cortical neurotoxicity ERK1 / ERK2 activation was from 82 to 93 and 96%, respectively. Modified EBSS and largely prevented by inhibition of PKC, relatively mildly vehicles had little effect on apoptotic-like death (data not prevented by that of CaM kinase II, and combined
shown). inhibition of these two had an additive and complete
inhibitory effect. These results strongly indicate that in ERK1 / ERK2 activation a PKC-dependent pathway was mainly involved, a CaM kinase II-dependent pathway was
4. Discussion relatively mildly involved, and these two pathways were
inhibition of PTK and PKC. Therefore, PKC and the PTK significantly prevented the glutamate-induced apoptotic-might act on the same pathway largely contributing to like cell death and combined inhibition of CaM kinase II ERK1 / ERK2 activation. The exact relationship between (but not PKC or PTK) and ERK1 / ERK2 had a significant PTK and PKC is unclear. However, two pathways may additive effect. These results suggest that the ERK1 / ER-exist. One may be PKC–c-Src pathway. Schlaepfer et al. K2-mediated apoptotic-like death was mainly a down-has shown that PKC-mediated c-Src activity played a role stream event of PKC and PTK, but not CaM kinase II, in in integrin-induced ERK1 / ERK2 activation in NIH 3T3 glutamate-induced cortical neurotoxicity.
fibroblasts [40]. And the other may be PKC–PYK2 We have previously observed that ERK1 / ERK2 activa-pathway. Treatment of PC12 cells with agents that in- tion reverted to basal level at 3 h after glutamate exposure
21
creased intracellular Ca - or activated PKC-stimulated in cortical excitotoxicity [26]. Other studies also revealed PYK2 activity, and overexpression of PYK2 increased that PP1, PP2A, PP2B and PTP were involved in the ERK1 / ERK2 activity in these cells [29]. Thus, an im- reversion of ERK1 / ERK2 activation in some other cases
21
portant connection linking Ca , PKC, PYK2 and ERK1 / [34,35,38,43]. However, little is known about the upstream ERK2 may be present in some neuronal cells. cascade(s) of the reversion of ERK1 / ERK2 activation in CAMP-dependent protein kinase (PKA) has been shown excitotoxicity. In the present study, reversion of ERK1 / activated after NMDA-R [7], although there are numerous ERK2 activation was largely prevented by 300 nM okadaic
21
examples of antagonism and synergism between Ca and acid, supposed to inhibit both PP1 and PP2A [4], but not cAMP signaling. PKA has also been shown to interfere by 5 nM okadaic acid, supposed to inhibit only PP2A [4]. with ERK1 / ERK2 activation in many cell types, although Thus, PP1, but not PP2A, was mainly involved in rever-the interactions between cAMP and ERK1 / ERK2 sig- sion of ERK1 / ERK2 activation. Moreover, such reversion naling system are complex and variable [20]. However, our was also largely prevented by inhibition of PTP and results suggest that PKA be not involved in ERK1 / ERK2 combined inhibition of PP1 and PTP had no significant
activation in excitotoxicity. additive effect. Therefore, PP1 and PTP might act on the
Another possible mediator of ERK1 / ERK2 activation in same signaling pathway largely contributing to the rever-excitotoxicity is NOS. NOS-produced nitric oxide (NO) sion of ERK1 / ERK2 activation in excitotoxicity.
has been shown involved not only in glutamate-induced Since we have also showed that either PKC or PTK was ERK1 / ERK2 activation [49,50] but also in excitotoxicity mainly involved in ERK1 / ERK2-mediated apoptotic-like [2,36]. Unexpectedly, we did not observed any effect of death in glutamate-induced neurotoxicity, we further de-L-NNA, a NOS inhibitor, on ERK1 / ERK2 activation in termined the role of protein phosphatases in glutamate-glutamate-induced cortical neurotoxicity, although we induced apoptotic-like death. Our results showed that found that L-NNA provided significant neuroprotection inhibition of either PP 1 or PTP could not only prevent the against the glutamate-induced excitotoxicity. Our finding is reversion of ERK1 / ERK2 activation but also promote contrary to that by Yun et al. [50]. The basis for this glutamate-induced apoptosis. This would suggest that discrepancy is unclear, but presumably reflects differences either PP 1 or PTP be involved in the restrain of gluta-in culture conditions. We used a serum-free medium mate-induced neurotoxicity. Therefore, they might have an specific to neuron culture, while they used a serum-con- opposite effect to PKC or PTK on glutamate-induced taining medium. In fact, the time course of ERK1 / ERK2 apoptotic-like death through regulating ERK1 / ERK2 acti-activation in our study is also different from theirs. This vation. However, it is still unclear whether these phosphat-may also reflects the different mechanism in ERK1 / ERK2 ases act just the reverse action(s) of those kinases. activation. They found that ERK1 / ERK2 is not fully In conclusion, we have clearly shown that in glutamate-activated until 10 min after either NO, or NMDA, or induced cortical neurotoxicity ERK1 / ERK2 activation was glutamate treatment, while we found that ERK1 / ERK2 mainly mediated by NMDA-R-induced influx of
extracel-21
mechanisms for neuronal survival, differentiation, and plasticity?,
Acknowledgements
Neuron 16 (1996) 233–236.
[17] D.A. Fruman, C.B. Klee, B.E. Bierer, S.J. Burakoff, Calcineurin
The authors thank Professor Zhida Xu for excellent phosphatase activity in T lymphocytes is inhibited by FK 506 and assistance in assessment of the apoptotic-like death. cyclosporin A, Proc. Natl. Acad. Sci. USA 89 (1992) 3686–3690. [18] K. Fukunaga, E. Miyamoto, Role of MAP kinase in neurons, Mol.
Neurobiol. 16 (1998) 79–95.
[19] P. Gass, M. Kiessling, H. Bading, Regionally selective stimulation
References of mitogen activated protein (MAP) kinase tyrosine phosphorylation
after generalized seizures in the rat brain, Neurosci. Lett. 162 (1993) [1] T. Akiyama, J. Ishida, S. Nakagawa, H. Ogawara, S. Watanabe, N. 39–42.
Itoh, M. Shibuya, Y. Fukami, Genistein, a specific inhibitor of [20] S.S. Grewal, R.D. York, P.J.S. Stork, Extracellular-signal-regulated tyrosine-specific protein kinases, J. Biol. Chem. 262 (1987) 5592– kinase signalling in neurons, Curr. Opin. Neurobiol. 9 (1999)
5595. 544–553.
[2] A. Almeida, J.P. Golanos, J.M. Medina, Nitric oxide mediates [21] I. Hajimohammadreza, A.W. Probert, L.L. Coughenour, S.A. glutamate induced mitochondrial depolarization in rat cortical Borosky, F.W. Marcoux, P.A. Boxer, K.K. Wang, A specific inhibitor neurons, Brain Res. 816 (1999) 580–586. of calcium / calmodulin-dependent protein kinase-II provides
neuro-¨
[3] T. Arendt, M. Holzer, A. Grobmann, D. Zedlick, M.K. Bruckner, protection against NMDA- and hypoxia / hypoglycemia-induced cell Increased expression and subcellular translocation of the mitogen death, J. Neurosci. 15 (1995) 4093–4101.
activated protein kinase kinase and mitogen-activated protein kinase [22] C.S. Hill, R. Treisman, Transcriptional regulation by extracellular in Alzheimer’s disease, Neuroscience 68 (1995) 5–18. signals: mechanisms and specificity, Cell 80 (1995) 199–211. [4] C. Bialojan, A. Takai, Inhibitory effect of a marine-sponge toxin, [23] T. Honore, S.N. Davies, J. Drejer, E.J. Fletcher, P. Jacobsen, D.
okadaic acid, on protein phosphatases, specificity and kinetics, Lodge, F.E. Nielsen, Quinoxalinediones: potent competitive non-Biochem. J. 256 (1988) 283–290. NMDA glutamate receptor antagonists, Science 241 (1988) 701– [5] E. Bonfoco, D. Krainc, M. Ankarcrona, P. Nicotera, S.A. Lipton, 703.
Apoptosis and necrosis: two distinct events induced, respectively, by [24] B.R. Hu, T. Wieloch, Tyrosine phosphorylation and activation of mild and intense insults with N-methyl-D-aspartate or nitric oxide / mitogen-activated protein kinase in the rat brain following transient superoxide in cortical cell cultures, Proc. Natl. Acad. Sci. USA 76 cerebral ischemia, J. Neurochem. 62 (1994) 1357–1367.
(1995) 7162–7166. [25] J. Ikeda, S. Terakawa, S. Murota, I. Morita, K. Hirakawa, Nuclear [6] G.J. Brewer, Serum-free B27 / neurobasal medium supports differen- disintegration as a leading step of glutamate excitotoxicity in brain
tiated growth of neurons from the striatum, substantia nigra, septum, neurons, J. Neurosci. Res. 43 (1996) 613–622.
cerebral cortex, cerebellum, and dentate gyrus, J. Neurosci. Res. 42 [26] Q. Jiang, Z. Gu, G. Zhang, G. Jin, Diphosphorylation and in-(1995) 674–683. volvement of extracellular signal-regulated kinases (ERK1 / 2) in [7] D.M. Chetkovich, J.D. Sweatt, NMDA receptor activation increases glutamate-induced apoptotic-like death in cultured rat cortical
cyclic AMP in area CA1 of the hippocampus via calcium / cal- neurons, Brain Res. 857 (2000) 71–77.
modulin stimulation of adenylyl cyclase, J. Neurochem. 61 (1993) [27] U.K. Laemmli, Cleavage of structural proteins during the assembly 1933–1942. of the head of bacteriophage T4, Nature 227 (1970) 680–685. [8] T. Chijiwa, A. Mishima, M. Hagiwara, M. Sano, K. Hayashi, T. [28] M. Leist, P. Nicotera, Calcium and neuronal death, Rev. Physiol.
Inoue, K. Naito, T. Toshioka, H. Hidaka, Inhibition of forskolin- Pharmacol. Biochem. 132 (1997) 79–125.
induced neurite outgrowth and protein phosphorylation by a newly [29] S. Lev, H. Moreno, R. Martinez, P. Canoll, E. Peles, J.M. Musac-synthesized selective inhibitor of cyclic AMP-dependent protein chio, G.D. Plowman, B. Rudy, J. Schlessinger, Protein tyrosine kinase, N-[2-( p-bromocinnamylamino)ethyl]-5-isoquinolinesul- kinase PYK2 involved in Ca (21)-induced regulation of ion channel fonamide (H-89), of PC12D pheochromocytoma cells, J. Biol. and MAP kinase functions, Nature 376 (1995) 737–745.
Chem. 265 (1990) 5267–5272. [30] S.A. Lipton, P.A. Rosenberg, Excitatory amino acids as a final [9] D.W. Choi, Calcium: still center-stage in hypoxic-ischemic neuronal common pathway for neurologic disorders, New Engl. J. Med. 330
death, Trends Neurosci. 18 (1995) 58–60. (1994) 613–622.
[10] R.J. Davis, The mitogen-activated protein kinase signal transduction [31] O.H. Lowry, H.J. Rosebrough, A.L. Farr, R.J. Randall, Protein pathway, J. Biol. Chem. 268 (1993) 14553–14556. measurement with Folin-phenol reagent, J. Biol. Chem. 193 (1951) [11] M.A. Dwyer, D.S. Bredt, S.H. Snyder, Nitric oxide synthase: 265–275.
irreversible inhibition byL-NG-nitroarginine in brain in vitro and in [32] C.J. Marshall, Specificity of receptor tyrosine kinase signaling: vivo, Biochem. Biophys. Res. Commun. 176 (1991) 1136–1141. transient versus sustained extracellular signal-regulated kinase acti-[12] M. Faucher, N. Girones, Y.A. Hannun, R.M. Bell, R.J. Davis, vation, Cell 80 (1995) 179–185.
Regulation of the epidermal growth factor receptor phosphorylation [33] S. Nakielny, P. Cohen, J. Wu, T. Sturgill, MAP kinase activator form state by sphingosine in A431 human epidermoid carcinoma cells, J. insulin-stimulated skeletal muscle is a protein threonine / tyrosine Biol. Chem. 263 (1988) 5319–5327. kinase, EMBO J. 11 (1992) 2123–2129.
[13] M. Favaron, H. Manev, R. Siman, M. Bertolino, A.M. Szekely, G. [34] E.D. Norman, E. Thiels, G. Barrionuevo, E. Klann, Long-term DeErausquin, A. Guiditti, E. Costa, Down-regulation of protein depression in the hippocampus in vivo is associated with protein kinase C protects cerebellar granule neurons in primary culture from phosphatase-dependent alterations in extracellular signal-regulated glutamate-induced neuronal death, Proc. Natl. Acad. Sci. USA 87 kinase, J. Neurochem. 74 (2000) 192–198.
(1990) 1983–1987. [35] M. Ogata, M. Oh-hora, A. Kosugi, T. Hamaoka, Inactivation of [14] V. Felipo, M.D. Minana, S. Grisolia, Inhibitors of protein kinase C mitogen-activated protein kinases by a mammalian tyrosine-specific prevent the toxicity of glutamate in primary neuronal cultures, Brain phosphatase, PTPBR7, Biochem. Biophys. Res. Commun. 256
Res. 604 (1993) 192–196. (1999) 52–56.
[15] I. Figiel, L. Kaczmarek, Orthovanadate induces cell death in rat [36] T. Ohmori, Y. Hirashima, M. Kurimoto, S. Endo, A. Takaku, In vitro dentate gyrus primary culture, Neuroreport 8 (1997) 2465–2470. hypoxia of cortical and hippocampal CA1 neurons: glutamate, nitric
21
selective neural death in CA1 neurons, Brain Res. 743 (1996) [43] J. Tian, M. Karin, Stimulation of Elk1 transcriptional activity by 109–115. mitogen-activated protein kinases is negatively regulated by protein [37] T. Ohtsuki, M. Matsumoto, K. Kitagawa, T. Mabuchi, K. Mandai, phosphatase 2B (calcineurin), J. Biol. Chem. 274 (1999) 15173–
K. Matsushita, K. Kuwabara, M. Tagaya, S. Ogawa, H. Ueda, T. 15180.
Kamada, T. Yanagihara, Delayed neuronal death in ischemic hip- [44] E.C. Toescu, Activity of voltage-operated calcium channels in rat pocampus involves stimulation of protein tyrosine phosphorylation, cerebellar granule neurons and neuronal survival, Neuroscience 94 Am. J. Physiol. 271 (1996) C1085–1097. (1999) 561–570.
[38] N. Parameswaran, P. Nambi, C.S. Hall, D.P. Brooks, W.S. Spielman, [45] H. Tokumitsu, T. Chijiwa, M. Hagiwara, A. Mizutani, M. Terasawa, Adrenomedullin decreases extracellular signal-regulated kinase ac- H. Hidaka, KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L -tivity through an increase in protein phosphatase-2A ac-tivity in tyrosyl]-4-phenylpiperazi, J. Biol. Chem. 265 (1990) 4315–4320. mesangial cells, Eur. J. Pharmacol. 388 (2000) 133–138. [46] H. Towbin, T. Stachelin, J. Gordon, Electrophoretic transfer of [39] G. Rusanescu, H. Qi, S.M. Thomas, J.S. Brugge, S. Halegoua, proteins from polyacrylamide gels to nitrocellulose sheets. Pro-Calcium influx induces neurite growth through a Src-Ras signaling cedure and some applications, Proc. Natl. Acad. Sci. USA 76 (1979) cassette, Neuron 15 (1995) 1415–1425. 4350–4354.
[40] D.D. Schlaepfer, K.C. Jones, T. Hunter, Multiple Grb2-mediated [47] S.R. Vincent, M. Sebben, A. Dumuis, J. Bockaert, Neurotransmitter integrin-stimulated signaling pathways to ERK2 / mitogen-activated regulation of MAP kinase signaling in striatal neurons in primary protein kinase: summation of both c-Src- and focal adhesion kinase- culture, Synapse 29 (1998) 29–36.
initiated tyrosine phosphorylation events, Mol. Cell. Biol. 18 (1998) [48] A.M. Wood, K.R. Bristow, N-Methyl-D-aspartate receptor desensiti-2571–2585. sation is neuroprotective by inhibiting glutamate-induced apoptotic-[41] D.D. Schoepp, B.G. Johnson, E.C. Smith, L.A. McQuaid, like death, J. Neurochem. 70 (1998) 677–687.
Stereoselectivity and mode of inhibition of phosphoinositide-cou- [49] H.Y. Yun, V.L. Dawson, T.M. Dawson, Glutamate-stimulated cal-pled excitatory amino acid receptors by 2-amino-3-phosphonop- cium activation of Ras / Erk pathway mediated by nitric oxide, ropionic acid, Mol. Pharmacol. 38 (1990) 222–228. Diabetes Res. Clin. Pract. 45 (1999) 113–115.
[42] R. Seger, N.G. Ahn, J. Posada, E.S. Munar, A.M. Jensen, J.A. [50] H.Y. Yun, M. Gonzalez-Zulueta, V.L. Dawson, T.M. Dawson, Nitric Cooper, M.H. Cobb, E.G. Krebs, Purification and characterization of oxide mediates N-methyl-D-aspartate receptor-induced activation of mitogen-activated protein kinase activator(s) from epidermal growth p21ras, Proc. Natl. Acad. Sci. USA 95 (1998) 5773–5778. factor-stimulated A431 cells, J. Biol. Chem. 267 (1992) 14373– [51] C.F. Zorumski, J.W. Olney, Excitotoxic neuronal damage and