Photosystem II evolves oxygen by using water in the unlikely role of a reductant. The absorption of sunlight by chlorophyll produces highly oxidizing equivalents that are filled with electrons stripped from water. This proton-coupled redox chemistry occurs at the oxygen-evolving complex, which contains a tetramanganese cluster, a redox-active tyrosine amino acid hydrogen-bonded to a histidine amino acid, a calcium ion and chloride. Hydrogen-atom abstraction by the tyrosyl radical from water bound to the manganese cluster is now widely held to occur in this process, at least for some of the steps in the catalytic cycle. We discuss kinetic and energetic constraints on the hydrogen-atom abstraction process.
Addresses
*Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, USA
†Department of Biochemistry, Arrhenius Laboratories for Natural Sciences, Stockholm University, S106 91 Stockholm, Sweden †Current address: Department of Biochemistry, Stockholm University, S-106 91 Stockholm, Sweden
Current Opinion in Plant Biology2000, 3:236–242 1369-5266/00/$ — see front matter
© 2000 Elsevier Science Ltd. All rights reserved.
Abbreviations ε
ε dielectric constant ∆
∆G free energy change
OEC oxygen-evolving complex
PSII photosystem II
QA initial plastoquinone electron acceptor in photosystem II
Sn State at which n oxidizing equivalents are accumulated (where n is 0—4)
Yz redox-active tyrosine at position 161 of the D1 protein
Introduction
Photosynthetic organisms use photosystem II (PSII) to cat-alyze the light-driven oxidation of water according to the following half-cell reaction:
2 H2O→O2+ 4 H++ 4 e–
The protons and electrons released in this process are ulti-mately used in starch production; to the plant, O2is simply a waste product. This remarkable redox chemistry is initi-ated by photon absorption by the PSII reaction-center chlorophyll, P680, which produces the charge-separated state, P680+QA–. Reduction of P680+ by water is mediated by the oxygen-evolving complex (OEC), which is largely, if not exclusively, associated with D1, one of the principal reaction-center proteins in PSII. The OEC consists of four manganese ions, a redox-active tyrosine called Yz(tyrosine 161), histidine 190, and Ca2+ and Cl– ions. The cluster con-taining the four Mn ions [i.e. (Mn)4] stores oxidizing
equivalents upon photon absorption; this process is anno-tated by the Snnomenclature, where n is the number of oxidizing equivalents accumulated. Four successive light-induced charge separations lead to the formation of S4from which O2 is released, thereby resetting the system to S0 (for recent reviews see [1••,2,3]).
It has been postulated that Yz•accepts both electrons and protons from water during catalysis [4,5], and a mechanism for the water-splitting chemistry of PSII based on H-atom abstraction has been developed [1••,6]. Several functional models have been proposed recently within this theme. These models differ primarily in the extent to which Yz acts as a H-atom abstractor during the catalytic cycle and in the O=O bond-forming step [7•–9•,10••]. In this review, we discuss the mechanistic details of coupled electron–proton transfer. First, we describe the original H-atom abstraction model, then we discuss Yzoxidation briefly, and finally we analyze Yz•reduction.
The hydrogen-atom abstraction model
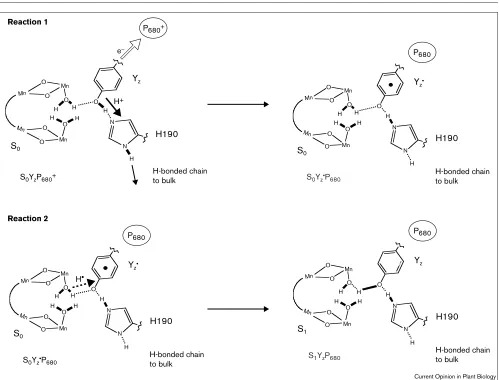
Essential features of this mechanism are summarized in Figure 1. Upon photon absorption and generation of P680+, Y
zreduces P680+and simultaneously deprotonates through the H190–H-bonded network, which is postulat-ed to extend to bulk, to produce Yz• (reaction 1 in Figure 1). In the second step, Yz• abstracts a hydrogen atom from the substrate water/hydroxide that is ligated to the (Mn)4cluster to advance the S-state (Figure 1, reac-tion 2). Four consecutive photon absorpreac-tion/H-atom abstraction events strip four hydrogen atoms from two substrate water molecules; the two oxygen atoms collapse to form O2, which is released as the system resets to the S0state by loading the next two water molecules. Within this mechanism, each cofactor in the OEC plays specific roles that are described below.
Yzcarries out at least three functions that are critical to the oxidation of water. First, Yz reduces P680+ on the ns time scale, easily outcompeting the P680+QA– charge-recombination reaction, which prevents energy loss as heat. Because of this rapid reduction of P680+, PSII operates with a quantum efficiency that is close to unity. Second, the oxidation of water requires strongly oxidizing conditions. These are initially created at P680+ and preserved by the high reduction potential of the Yz•/Yzredox couple. Third, Yz interfaces the electron-tunneling reactions that are carried out by the P680/QApair at the reaction center to the proton-coupled redox chemistry in the OEC. Thus, the proton currents associated with water oxidation are switched on at Yz•.
Concerted hydrogen-atom abstraction in photosynthetic
water oxidation
Kristi L Westphal
*
, Cecilia Tommos
†
, Robert I Cukier
*
and
Site-directed mutagenesis ([11••] and references therein) and kinetic data [12•,13•] have shown that Y
zis hydrogen bonded to H190. If this histidine is mutated, Yzoxidation slows and takes ms rather than ns. Furthermore, these results suggest that when hydrogen-bond formation is cur-tailed, Yzcannot deprotonate efficiently and is not able to reduce P680+. Thus, the oxidation of Yzis a proton-coupled event, and H190 serves as the initial proton acceptor. The electron made available in this process reduces P680+, and the proton migrates through the H190/H-bonded network to bulk ([14•,15••], but see [10••]). Together, these processes form the neutral radical, Yz•, which is now able to mediate the H-atom abstractions.
Yz•and the (Mn)4cluster, which have a magnetic center-to-center separation of about 8 Å ([16•] and references within), form a single catalytic site. Within this complex,
the function of the (Mn)4 cluster is at least three-fold. First, it serves to delocalize the oxidizing equivalents gen-erated as the S-state advances. The general consensus of opinion is that the (Mn)4cluster is oxidized on each S-state transition, with the possible exception of the S2→S3 transi-tion [17,18]. Second, it reduces the bond-dissociatransi-tion energy of bound substrate [1••,19,20•]. Third, the (Mn
4) cluster acts as a template for the formation of the O=O bond [6]. The C-shaped structure promoted by Klein, Sauer, Yachandra and their co-workers [21] is ideal in this regard, as it holds the two oxygen atoms in a geometry that allows facile O=O bond formation. The structure of the (Mn)4cluster is controversial but a templating function of the metal cluster is likely.
Ca2+and Cl– are closely associated with the catalytic site [21]. We have proposed that Ca2+serves as a Cl–docking Figure 1
to bulk H-bonded chainto bulk
Yz Yz•
Current Opinion in Plant Biology Reaction 1
Reaction 2
Essential features of the H-atom abstraction model for photosynthetic water oxidation. The postulated relative arrangement of the catalytic center of PSII is shown. Substrate water binds terminally to the Mn ions within hydrogen bonding (……) distance of the Yzphenol group. The arrows show the flow of electrons (open arrow) and protons
(solid arrow) on the proton-coupled oxidation of Yz(Reaction 1) and on the subsequent H-atom transfer (broken arrow) to Yz• from water
site, and that Cl–neutralizes charge upon the S
2→S3 tran-sition as the result of Jahn-Teller effects [1••]. The role of these two essential ions is elusive, however, and several other functions have been suggested (e.g. [7•–9•]).
There is considerable experimental support for H-atom abstraction in the catalytic cycle of PSII. In the sections that follow, we provide further insight into the mechanisms of H-atom transfer from and to Yzduring water oxidation.
The kinetics of Y
zoxidation
The oxidation of Yz has S-state dependent, multiphasic electron-transfer kinetics that occur in the ns to µs
timescale. Moreover, its oxidation rate is highly sensitive to the integrity of the OEC (Figure 2). The different kinetics for Yz oxidation in holo- versus apo-PSII have recently been considered in detail experimentally [11••,12•,13•,22,23] and in a review article [15••]. In the intact system (i.e. holo-PSII), it appears that proton trans-fer is facilitated by a well-ordered H-bonding structure (Figure 2a). Under these conditions, proton transfer is not rate limiting and the kinetics of Yzoxidation can be accu-rately predicted [15••] using an expression developed by Dutton, Moser, and coworkers for estimating the rates of intraprotein, non-adiabatic electron tunneling [24••]. When PSII is depleted of Mn or Ca (i.e. apo-PSII), hydro-gen bonding at the Yzsite becomes disordered (Figure 2b). Under these conditions, the deprotonation of Yz during electron transfer is no longer facile and the rate of the pro-ton transfer event limits that of Yzoxidation. The overall reaction slows to the µs timescale and becomes pH
depen-dent with a substantial kinetic isotope effect. These advances in our understanding of the experimental details of Yzoxidation have been accompanied by a satisfying the-oretical description of the process as an example of dissociative proton-coupled electron transfer [25•].
The kinetics of Y
zreduction
The reduction of Yz• is S-state dependent and occurs over the µs to ms time scale (Table 1). Assuming reason-able values for driving force, reorganization energy, protein packing and distance, we find that the expres-sion developed by Page et al. [24••] predicts electron-tunneling rates that are far more rapid than the observed rates of Yz•reduction [15••]. Yzdeprotonates to H190 upon oxidation and reprotonates upon reduction. It follows that an accurate analysis of Yz• reduction should take into account the movement of a proton as well as an electron. The Yz•/Yz and Sn+1/Sn reduction potentials, and the activation energies and pre-exponen-tial factors for the S-state transitions, are required for this analysis and are given in Table 2.
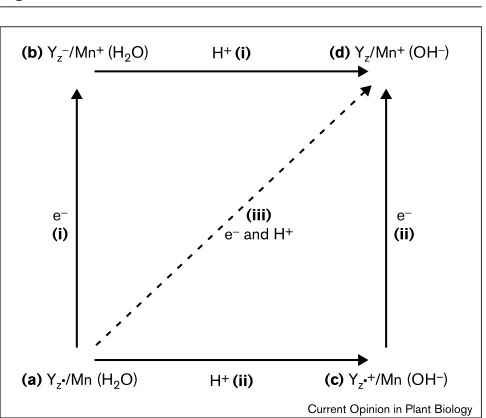
Three mechanisms for the transfer of an electron and a proton are illustrated in Figure 3, where the Sn→Sn+1 advance is represented by Mn(H2O)→Mn+(OH-). Two are sequential and involve either electron transfer fol-lowed by proton transfer [path (i)], or proton transfer followed by electron transfer [path (ii)]. The third mech-anism invokes concerted electron/proton transfer [path (iii)].
Figure 2
Rate-limiting factors for Yzoxidation. (a)The OEC is intact, proton transfer to H190 occurs on the ns time scale or faster, and the reduction of P680+is limited by the rate of
electron transfer. (b)PSII is depleted of Mn or Ca and the oxidation of Yz takes µs rather than
ns. The movement of the phenol proton now becomes rate-limiting for the overall proton-coupled reduction of P680+.
CH2
CH2 CH2
CH2
O H
N
N H
fast
e−
slow P680+
O H
N
N
H
(a) Holo-PSII (ns kinetics)
(b) Apo-PSII (µs kinetics)
Well-ordered Disordered
fast e−
P680+
H190 H190
H+
slowH+
Current Opinion in Plant Biology
Table 1
Half-times for S-state advance..
S-state Thylakoids PSII Core
membranes particles Yz•S0→YzS1 40–60 µs 30–70 µs 60 µs
Yz•S1→YzS2 65–86 µs 30–70 µs 75–95 µs
Yz•S2→YzS3 140–245 µs 55–110 µs 225–380 µs
Yz•S3→YzS0 750 µs;1.3 ms 1.2–1.4 ms 4.1–4.6 ms
For path (i), the initial transfer of an electron results in the reduction of Yz•to a tyrosinate [intermediate (b)], which subsequently protonates in the second step of the process. The Yz•/Yzpotential (shown in Table 2), corresponds to the overall H+/e– transfer, but to judge the viability of the sequential process we must consider the energetics of forming the transient intermediate (b). The potential of Y•/Y–is 0.68 V in water [15••] and is expected to decrease in the low protein dielectric. This effect can be estimated by using the Born model [26]:
∆G = 14.397/εr (1)
where ∆G (in units of volts) is the electrostatic penalty for
introducing a charged species with radius (r; in Å) in a homogenous medium of dielectric constant ε. Assuming a
radius of 3 Å and an εof 10, we calculate a Yz•/Yz–potential of 0.26 V. Thus, in a protein milieu, Yz•is a relatively weak oxidant if Yz–is the product. For the Sn+1/Snpotential, the opposite trend occurs, that is, to oxidize [Mn–H2O] or [Mn–OH] to charged products requires a stronger oxidant than when the process is proton coupled. With εand r as
used above for Yz, Equation 1 predicts that the S1/S0 potential increases from 0.7 V to 1.12 V, if a positive charge is formed. For the higher S-state transitions, correspond-ingly higher reduction potentials are predicted. As a result, the driving force for forming (b) during S0→S1 is very endergonic, (+0.26)–(+1.12) = –0.86 V. The electrostatic interaction between [Mn-OH2]+and Yz–in (b) lowers this value by –0.18 V. Thus, the resulting difference in reduc-tion potential is –0.68 V, which equates to a free energy difference (∆G) of 15.7 kcal/mol. For the higher S-states,
the endergonicity will be even greater. These free energy values obtained for formation of (b) lead to activation ener-gies that are substantially higher than the activation energies for the S-state transitions presented in Table 2, and suggest that path (i) in Figure 3 is improbable.
The second possibility is also sequential [path (ii) in Fig-ure 3]. The pKas of the proton donor and acceptor can be used to determine the energetics of the initial proton trans-fer. For the individual S-states, the pKais expected to be greater than or equal to seven [1••,20•], whereas the pK
aof Y•+ in water is minus two [15••]. Therefore, the ∆pK
afor the proton transfer is at least nine, resulting in a ∆G of at
least 12 kcal/mol. The ∆G is as high as 23 kcal/mol when
Equation 1 is used to estimate the pKain a protein envi-ronment. Consequently, the ∆G calculated for the transfer
of a proton prior to electron transfer gives activation ener-gies that are considerably higher than those that have been observed for any of the S-state transitions (Table 2). We conclude that reduction of Yz•is unlikely to take place by this mechanism.
As the sequential transfer of an electron and a proton involves the formation of high energy intermediates, con-certed proton/electron transfer in PSII becomes a viable mechanism. These processes have been studied theoreti-cally by Cukier [25•,27••], and by Hammes-Schiffer [28], and experimentally by researchers including Mahoney and DaRooge [29], Ingold and colleagues [30,31], Nocera and Figure 3
Possible reaction paths for electron and proton transfer. Four species are shown as follows: (a) The reactant state Yz•/Mn(H2O), (d)the product state Yz/Mn+(OH-), and two intermediate states, (b) Yz
-/Mn+(H2O) and (c) Yz•+/Mn(OH-). Along the edges of the square, two
sequential pathways are shown. In path (i), electron transfer is followed by proton transfer (et/pt), while in path (ii), the reverse sequence of proton and electron steps occurs (pt/et). In path (iii) the electron and proton move in a concerted process.
(b) Yz−/Mn+ (H2O)
(a) Yz•/Mn (H2O)
(iii)
e− and H+
H+ (i)
H+ (ii)
e− (ii)
e− (i)
(d) Yz/Mn+ (OH−)
(c) Yz•+/Mn (OH–)
Current Opinion in Plant Biology
Table 2
Kinetic and thermodynamic parameters for the S-state transitions..
Reaction Ea(kcal/mol) A (s–1) KIE Yz•/Yz(V) Sn+1/Sn
Yz•S0→YzS1 1.2 0.97 0.70
Yz•S1→YzS2 3.0 4.0 ×106 1.3; 2.9 0.97 0.93
Yz•S2→YzS3 9.0 5.4 ×109 1.3 0.97 0.93
Yz•S3→YzS0 5.0† 8.9 ×105† 1.4–1.6 0.97 0.93
9.2‡ 2.9 ×1014‡
colleagues [27••,32], Mayer and colleagues [33••,34], and Sjödin et al. [35]. Concerted processes are expected to be unlikely,a priori, as they require simultaneous tunneling of the electron and proton. The proton contributes to the pro-ton-coupled electron transfer rate through Franck-Condon factors whose magnitudes can be quite small in view of the large displacement of the proton upon transfer. Cukier refers to this as ‘Franck-Condon drag’. Thus, coupled elec-tron-transfer/proton-transfer processes are inherently low probability events, and are associated with substantially negative activation entropies, and hence smaller Arrhenius pre-exponential factors (A), that are a hallmark of atom transfers. These Avalues, when converted to s–1units, as appropriate to unimolecular reactions, are around 108.5s–1 [29,36]. These values are significantly lower than the 1013 s–1that is taken for van der Waal’s electron transfer [24••]. In essence, then, there is a competition between concerted and sequential routes (Figure 3). A sequential process may be slow because of a rate-limiting endergonic step, where-as the concerted process may be slow because of Franck-Condon limitations. If the sequential process pro-ceeds through an intermediate [in the specific case of PSII, intermediates (b) or (c) in Figure 3] that is accessible ther-mally, then the sequential route will dominate [24••]. Conversely, if the sequential intermediates lie too high in energy, as they appear to in PSII, then the concerted path-way dominates (Figure 4).
From model compound studies, general characteristics of concerted electron and proton transfer reactions can be
extracted. The activation energies of these processes are generally very low. For example, activation energies in the 1.7–4.9 kcal/mol range were reported in studies on the reactivity of phenoxy radicals in H-atom abstraction reac-tions [29,30] and values of 5 kcal/mol were measured for H-atom transfer between two ferric complexes [34]. The activation energies measured for reduction of Yz• are 1–9 kcal/mol (Table 2) and fit well within the range of val-ues for H-atom abstraction reactions.
Similarly, moderate kinetic isotope effects, typically less than 2.5, and reaction rates in the range 103–106s–1, despite short distances, are typical for atom abstraction processes [25•,27••,29–31,33••,34]. These results are well in line with those that have been obtained for PSII (Table 2). Thus, there is good agreement between the kinetic characteristics of S-state advance and observations of H-atom abstraction in model systems. Moreover, the above analysis indicates that sequential processes proceed through intermediates that are inaccessible energetically. Accordingly, it appears that the reduction of Yz•involves the concerted movement of a proton and an electron from the Mn–water (hydroxide) complex on each S-state transition.
Conclusions
The original model for H•atom abstraction as the underly-ing mechanism for water oxidation in PSII stressed the necessity of managing both proton and electron currents closely [4,37]. The mutagenesis and kinetic results for Yz oxidation by P680+obtained in the past two years reinforce this point: PSII in the absence of either H190 or the (Mn)4 cluster is not an efficient oxidant of Yz. The dramatic kinet-ic slow-downs of Yzoxidation rate when either H190 or the (Mn)4 cluster is missing are related to the extent that the local H-bond network around Yz is disrupted by their absence. The analysis presented above shows that the same principles apply for the reduction of Yz•. The available data indicate that protons and electrons move together in a con-certed process as the S-state advance occurs.
Acknowledgements
We would like to thank The Swedish Foundation for International Cooperation in Research and Higher Education, the US Department of Agriculture Competitive Grants Office, and NIH Grants GM-47274 and GM-37300 for financial support. We would also like to thank Chris Moser and Les Dutton for useful discussions along with Jim Mayer and Curt Hoganson for discussion and for careful reading of the manuscript.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
••of outstanding interest
1. Tommos C, Babcock GT: Oxygen production in nature: a light
•• driven metalloradical enzyme process.Accounts Chem Res1998,
31:18-25.
The authors summarize our knowledge of H-atom abstraction in the water-oxidation process. This information is complemented with a discussion of the
pKas, bond dissociation energies and local thermodynamic properties of
manganese-bound water. Figure 4
Potential energy surfaces showing the relation between the activation energies (Eas) for the sequential processes and for the concerted H-atom transfer. The Eas for the sequential processes are considerably higher than those for the concerted transfer (as detailed in the text). However, for the concerted process the pre-exponential (Afactor) is
significantly smaller than that for the sequential transfer. In PSII, therefore, the concerted process describes the S-state advance more accurately than either of the possible sequential routes.
Reaction Path
Energy
Yz•/Mn (H2O)
Yz/Mn+ (OH−) Concerted Sequential
Yz•+/Mn (OH−)
or Yz−/Mn+ (H2O)
2. Yocum CF, Pecoraro VL: Recent advances in the understanding of the biological chemistry of manganese.Curr Opin Chem Biol
1999, 3:182-187.
3. Barber J, Kühlbrandt W: Photosystem II.Curr Opin Struct Biol
1999, 9:469-475.
4. Hoganson CW, Lydakis-Simantiris N, Tang X-S, Tommos C, Warncke K,
Babcock GT, Diner BA, McCracken J, Styring S: A hydrogen-atom
abstraction model for the function of Yzin photosynthetic oxygen
evolution.Photosynth Res1995, 46:177-184.
5. Gilchrist ML Jr, Ball JA, Randall DW, Britt RD: Proximity of the
manganese cluster of photosystem-II to the redox-active tyrosine Yz.Proc Natl Acad Sci USA1995, 92:9545-9549.
6. Hoganson CW, Babcock GT: A metalloradical mechanism for the
generation of oxygen from water in photosynthesis.Science1997,
277:1953-1956.
7. Pecoraro VL, Baldwin MJ, Caudle MT, Hsieh W-Y, Law NA: A
• proposal for water oxidation in photosystem II.Pure Appl Chem
1998, 70:925-929.
The role of H-atom abstraction, manganese-cluster redox potentials, and the
possible involvement of Ca2+in presenting a hydroxide nucleophile close
enough to a putative Mnv=O species to form the critical 0=0 bond in the S
4
state is presented.
8. Limburg J, Vrettos JS, Liable-Sands LM, Rheingold AL, Crabtree RH,
• Brudvig GW: A functional model for O–O bond formation by the
O2-evolving complex in photosystem II.Science1999,
283:1524-1527.
A binuclear manganese center is described that promotes formation of the O=O bond. Tests of this mechanism using model complexes are summarized.
9. Siegbahn PEM, Crabtree RH: Manganese oxyl radical
• intermediates and O–O bond formation in photosynthetic oxygen
evolution and a proposed role for the calcium cofactor in photosystem II.J Am Chem Soc1999, 121:117-127.
The authors note the energetic difficulties involved in abstracting an H-atom from hydroxide that is ligated to a six-coordinate manganese ion. A mecha-nism to overcome this problem, which involves oxyl radical formation and
Ca2+involvement, is presented.
10. Haumann M, Junge W: Photosynthetic water oxidation: a simplex
•• scheme of its partial reactions.Biochim Biophys Acta1999,
1411:86-91.
An interpretation of proton release and chromophore bandshifts that are attributed to the formation of charge is presented that differs from the one described in this review. A complete model for water oxidation is presented that incorporates both charge-accumulating and charge-neutral transitions at the Mn complex.
11. Hays A-MA, Vassiliev IR, Golbeck JH, Debus RJ: Role of D1-His190 in
•• the proton-coupled oxidation of tyrosine Yzin
manganese-depleted photosystem II.Biochemistry1999, 38:11851-11865. Mutations at H190, with and without chemical rescue by exogenous bases, are used to establish this central role of the histidine in promoting
deproto-nation of Yzas it is oxidized by P680+.
12. Schilstra MJ, Rappaport F, Nugent JHA, Barnett CJ, Klug DR:
• Proton/hydrogen transfer affects the S-state-dependent
microsecond phases of P680+reduction during water splitting.
Biochemistry1998, 37:3974-3981.
The role of proton transfer in facilitating the slower phases of P680+
reduc-tion that involve proton/hydrogen moreduc-tion in water-splitting PSII preparareduc-tions is described.
13. Christen G, Seeliger A, Renger G: P680+*reduction kinetics and
• redox transition probability of the water oxidizing complex as a
function of pH and H/D isotope exchange in spinach thylakoids.
Biochemistry1999, 38:6082-6092.
The authors show that both proton transfer events and deuterium kinetic
iso-tope effects are apparent in the slower phases of P680+reduction in
water-splitting PSII preparations. This behavior contrasts with that exhibited during
the ns time-scale phases of P680+reduction in the same preparations and
leads to the development of the model described in [15••].
14. Tommos C, Hoganson CW, Di Valentin M, Lydakis-Simantiris N,
• Dorlet P, Westphal K, Chu H-A, McCracken J, Babcock GT:
Manganese and tyrosyl radical function in photosynthetic oxygen evolution.Curr Opin Chem Biol1998, 2:244-252.
The H-atom transfer model is discussed and interpretations of elec-trochromism that do not involve charge acccumulations are considered.
15. Tommos C, Babcock GT: Proton and hydrogen currents in
•• photosynthetic water oxidation.Biochim Biophys Acta2000, in press.
This review considers the oxidation of tyrosine in aqueous solution and of Yzby
P680+. It summarizes results that support the model in Figure 2 for the oxidation
process and shows that the reduction of Yz• during the S-state advances
can-not be explained by simple electron transfer.
16. Lakshmi KV, Eaton SS, Eaton GR, Brudvig GW: Orientation of the
• tetranuclear manganese cluster and tyrosine Z in the O2-evolving
complex of photosystem II: an EPR study of the S2Yz*state in
oriented acetate-inhibited photosystem II membranes.
Biochemistry1999, 38:12758-12767.
The proximity of Yzand the (Mn)4cluster is demonstrated by using electron
paramagnetic resonance (EPR) spectroscopy on oriented samples; earlier work using EPR that reached the same conclusion is summarized. 17. Roelofs TA, Liang WC, Latimer MJ, Cinco RM, Rompel A,
Andrews JC, Sauer K, Yachandra VK, Klein MP: Oxidation states of
the manganese cluster during the flash-induced S-state cycle of the photosynthetic oxygen-evolving complex.Proc Natl Acad Sci
USA1996, 93:3335-3340.
18. Iuzzolino L, Dittmer J, Dorner W, Meyer-Klaucke W, Dau H: X-ray
absorption spectroscopy on layered photosystem II membrane particles suggests manganese-centered oxidation of the oxygen-evolving complex for the S0–S1, S1–S2, and S2–S3transitions of
the water oxidation cycle.Biochemistry1998, 37:17112-17119. 19. Blomberg MRA, Seigbahn PEM, Styring S, Babcock GT, Åkermark B,
Korall P: A quantum chemical study of hydrogen abstraction from
manganese-coordinated water by a tyrosyl radical: a model for water oxidation in photosystem II.J Am Chem Soc1997,
119:8285-8292.
20. Law NA, Caudle MT, Pecoraro VL: Manganese redox enzymes and
• model systems: properties, structures, and reactivity.Adv Inorg
Chem1999, 46:305-440.
This general review of manganese function in biology discusses photosyn-thetic water-oxidation in depth.
21. Cinco RM, Robblee JH, Rompel A, Fernandez C, Yachandra VK,
Sauer K, Klein MP: Strontium EXAFS reveals the proximity of
calcium to the manganese cluster of oxygen-evolving photosystem II.J Phys Chem1998, 102:8248-8256.
22. Diner BA, Force DA, Randall DW, Britt RD: Hydrogen bonding, solvent
exchange, and coupled proton and electron transfer in the oxidation and reduction of redox-active tyrosine Yzin Mn-depleted core
complexes of photosystem II.Biochemistry1998, 37:17931-17943.
23. Tommos C, McCracken J, Styring S, Babcock GT: Stepwise
disintegration of the photosynthetic oxygen-evolving complex.
J Am Chem Soc1998, 120:10441-10452.
24. Page CC, Moser CC, Chen X, Dutton PL: Natural engineering
•• principles of electron tunneling in biological oxidation-reduction. Nature1999, 402:47-52.
This paper provides a seminal discussion of electron transfer in biological systems. The role of endergonic steps in an overall exergonic electron trans-fer process is considered.
25. Cukier RI: A theory for the rate constant of a dissociative proton
• coupled electron-transfer reaction.J Phys Chem A1999,
103:5989-5995.
Dissociative proton-coupled electron transfer, which is typified by the
oxida-tion of Yzby P680+in PSII, is analysed in considerable theoretical depth.
26. Rich PR, Meunier B, Mitchell R, Moody AJ: Coupling of charge and
proton movement in cytochrome coxidase.Biochim Biophys Acta
1996, 1275:91-95.
27. Cukier RI, Nocera DG: Proton-coupled electron transfer.Annu Rev
•• Phys Chem1998, 49:337-369.
The theory of coupled proton/electron transfer in both chemistry and biolo-gy is summarized and selected applications are presented.
28. Soudackov A, Hammes-Schiffer S: Multistate continuum theory for
multiple charge transfer reactions in solution.J Chem Phys1999,
111:4672-4687.
29. Mahoney LR, DaRooge MA: The kinetic behavior and
thermochemical properties of phenoxy radicals.J Am Chem Soc
1975, 97:4722-4731.
30. Foti M, Ingold KU, Lusztyk J: The surprisingly high reactivity of
phenoxy radicals.J Am Chem Soc1994, 116:9440-9447.
31. MacFaul PA, Ingold KU, Lusztyk J: Kinetic solvent effects on
hydrogen atom abstraction from phenol, aniline, and diphenylamine. The importance of hydrogen bonding on their radical-trapping (antioxidant) activities.J Org Chem1996,
32. Kirby JP, van Dantzig NA, Chang CK, Nocera DG: Formation of porphryn donor-acceptor complexes via an amidinium-carboxylate salt bridge.Tetrahedron Lett1995, 36:3477-3480.
33. Mayer JM: Hydrogen atom abstraction by metal-oxo complexes:
•• understanding the analogy with organic radical reactions. Accounts Chem Res1998, 31:441-450.
The basic thermodynamic and kinetic principles, such as low activation ener-gies and low rates of H-atom abstraction by metal-oxo complexes, are pre-sented. These are compared to analogous processes in which the H-atom acceptor is an organic moiety.
34. Roth JP, Lovell S, Mayer JM: Intrinsic barriers for electron and
hydrogen atom transfer reactions of biomimetic iron complexes.
J Am Chem Soc 2000, 122:in press.
35. Sjödin M, Styring S, Åkermark B, Sun L, Hammarström L:
Proton-coupled electron transfer from tyrosine in a tyrosine-ruthenium-tris-bipyridine complex; comparison with tyrosinezoxidation in
photosystem II. J Am Chem Soc 2000, 122:in press.
36. Benson SW: Thermochemical Kinetics: Methods for The Estimation
of Thermochemical Data And Rate Parameters, edn 2. New York: Wiley; 1976.
37. Tommos C, Tang X-S, Warncke K, Hoganson CW, Styring S,
McCracken J, Diner BA, Babcock GT: Spin-density distribution,
conformation, and hydrogen-bonding of the redox-active tyrosine Yzin photosystem-II from multiple electron magnetic resonance
spectroscopies — implications for photosynthetic oxygen evolution.J Am Chem Soc1995, 117:10325-10335.
38. Karge M, Irrgang K-D, Renger G: Analysis of the reaction coordinate
of photosynthetic water oxidation by kinetic measurements of 355 nm absorption changes at different temperatures in photosystem II preparations suspended in either H2O or D2O.
Biochemistry1997, 36:8904-8913.
39. Razeghifard MR, Pace RJ: Electron paramagnetic resonance kinetic