Review article
Molecular mechanisms, lipoprotein abnormalities and
atherogenicity of hyperalphalipoproteinemia
Shizuya Yamashita
a,*, Takao Maruyama
a, Ken-ichi Hirano
a, Naohiko Sakai
a,
Norimichi Nakajima
b, Yuji Matsuzawa
aaDepartment of Internal Medicine and Molecular Science,Graduate School of Medicine,B5,Osaka Uni6ersity,2-2Yamada-oka,Suita,
Osaka565-0871,Japan
bNakajima Clinic,Omagari,Akita014-0013,Japan
Received 19 February 1999; received in revised form 11 May 2000; accepted 17 June 2000
Abstract
Hyperalphalipoproteinemia (HALP) is caused by a variety of genetic and environmental factors. Among these, plasma cholesteryl ester transfer protein (CETP) deficiency is the most important and frequent cause of HALP in the Asian populations. CETP facilitates the transfer of cholesteryl ester (CE) from high density lipoprotein (HDL) to apolipoprotein (apo) B-containing lipoproteins, and is a key protein in the reverse cholesterol transport system. The deficiency of CETP causes various abnormalities in the concentration, composition, and function of both HDL and low density lipoprotein (LDL). The significance of CETP in terms of atherosclerosis had been controversial. However, the in vitro evidence showed large CE-rich HDL particles in CETP deficiency are defective in cholesterol efflux. Similarly, scavenger receptor BI (SR-BI) knockout mice show a marked increase in HDL-cholesterol but accelerated atherosclerosis in atherosclerosis-susceptible mice. Recent epidemiological studies in Japanese – Americans and in Omagari area where HALP subjects with the intron 14 splicing defect of CETP gene are markedly frequent, have demonstrated an increased incidence of coronary atherosclerosis in CETP-deficient patients. Thus, CETP deficiency is a state of impaired reverse cholesterol transport which may possibly lead to the development of atherosclerosis. The current review will focus on the molecular mechanisms and atherogenicity of HALP, especially CETP deficiency. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Hyperalphalipoproteinemia; Cholesteryl ester transfer protein deficiency; Reverse cholesterol transport; Atherosclerosis
www.elsevier.com/locate/atherosclerosis
1. Introduction
Plasma high density lipoprotein (HDL)-cholesterol levels are negatively correlated with the incidence of coronary heart disease, suggesting a role for HDL in
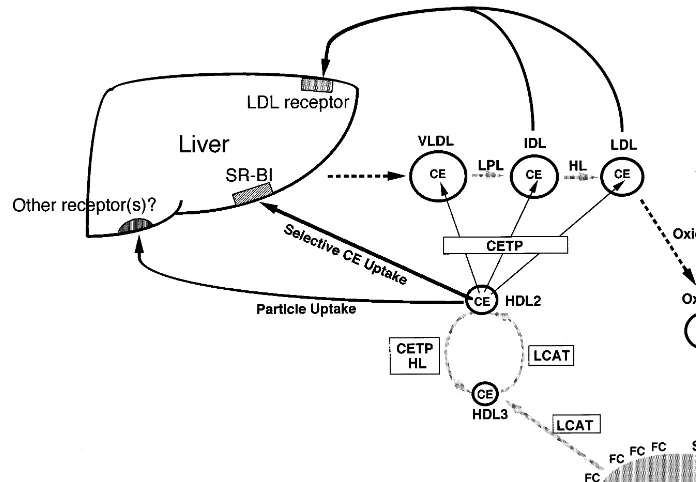
preventing the development of atherosclerosis. HDL takes up cholesterol from peripheral tissues and the cholesterol is esterified by lecithin:cholesterol acyltrans-ferase (LCAT). The cholesteryl ester (CE) is transferred by plasma cholesteryl ester transfer protein (CETP) to very low density lipoprotein (VLDL), intermediate den-sity lipoprotein (IDL) and low denden-sity lipoprotein (LDL) [1,2]. IDL and LDL are then catabolized via hepatic LDL receptor. The HDL obtains apolipo-protein (apo) E and apo E-containing HDL is taken up by the liver via LDL receptor or remnant receptor. Recent evidence shows that the CE moiety of HDL is selectively taken up by the liver via scavenger receptor class B type I (SR-BI) [3]. The HDL becomes enriched with triglycerides (TG) after the CETP-mediated trans-Abbre6iations: Apo, apolipoprotein; CE, cholesteryl ester; CETP,
cholesteryl ester transfer protein; HALP, hyperalphalipoproteinemia; HDL, high density lipoprotein; HL, hepatic lipase; IDL, intermediate density lipoprotein; LCAT, lecithin:cholesterol acyltransferase; LDL, low density lipoprotein; PBC, primary biliary cirrhosis; SR-BI, scav-enger receptor class B type I; TG, triglycerides; VHDL, very high density lipoprotein; VLDL, very low density lipoprotein.
* Corresponding author. Tel.: +81-6-68793738; fax: + 81-6-68793739.
E-mail address:[email protected] (S. Yamashita).
fer of CE, is hydrolyzed by hepatic lipase (HL) and gets smaller to take up cholesterol. Thus, HDL serves as a shuttle that tranports excess cholesterol from peripheral tissues to the liver for excretion. This pathway was designated as ‘reverse cholesterol transport’ system [4 – 6]. The HDL particles are heterogeneous and recent literature by in vitro experiments shows that preb -mi-grating HDL may have a very potent antiatherogenic function [7,8].
The first step of reverse cholesterol transport is the interaction between HDL and peripheral cells such as fibroblasts and macrophages, which is postulated to include aqueous diffusion, lipid-free apolipoprotein membrane microsolubilization, and SR-BI-mediated cholesterol flux [9 – 12]. Tangier disease is a genetic deficiency of HDL characterized by an absence of plasma HDL and deposition of CEs in the reticulo-en-dothelial system with splenomegaly and enlargement of tonsils and lymph nodes. HDL-mediated cholesterol efflux and intracellular lipid trafficking and turnover are abnormal in Tangier fibroblasts. Recent studies have demonstrated that Tangier disease is caused by mutations of ATP-binding cassette transporter 1 (ABC1) which may be involved in the efflux of choles-terol from cells [13 – 15].
Although low levels of serum HDL-cholesterol may lead to the development of atherosclerosis, there has been no consensus as to whether subjects with markedly high serum HDL-cholesterol levels are resis-tant to atherosclerosis. Hyperalphalipoproteinemia
(HALP) is a condition caused by various factors and is also associated with some diseases. Although familial HALP with hypobetalipoproteinemia described by Glueck et al. [16] was reported to be accompanied by longevity due to a low incidence of coronary heart disease, the data were not confirmed. Matsuzawa et al. reported two markedly hyperalphalipoproteinemic pa-tients with premature corneal opacity [17,18], which is a sign of lipid depositions in tissues. One of these patients was accompanied by angina pectoris. Therefore, these reports were the first descriptions suggesting that HALP may be a disorder of ‘reverse cholesterol trans-port’, which can be accompanied by atherosclerosis. These cases suggested that ‘dysfunctional HDL’ may be a potential mechanism leading to increased atheroscle-rosis despite high serum HDL-cholesterol levels.
2. Pathogenesis of HALP
HALP is associated with various diseases and caused by a variety of factors (Table 1). Some of these can be explained by mutations of molecules involved in HDL metabolism.
2.1. Primary HALP
The most important cause of primary HALP is a genetic deficiency of CETP, which will be discussed later. Groener et al. reported an HALP family with normal CETP activity [19], and the size and composi-tion of HDL particles of a case with HALP reported by Patsch et al. [20] were normal. Therefore, these cases may not be genetic CETP deficiency. Genetic deficiency of HL can also cause HALP. HL-deficient patients often develop HALP and coronary heart disease [21,22] and their HDL particles are enlarged and rich in TG [23]. In the two HALP cases with corneal opacity [18], HL activity was decreased.
Rader et al. reported a case of familial HALP who presented a markedly increased production rate of apo A-I [24]. The primary sequence of the proband’s apo A-I gene, including the 5%-flanking sequence, was nor-mal in this patient. Thus, the mechanism for the apo A-I overproduction in this case is unknown. Recently, a family with vertical transmission of simultaneous in-crease in HDL-cholesterol, LDL-cholesterol, apo A-I and apo B has been reported [25]. This condition was designated as familial HALP and hyperbetalipo-proteinemia. The affected patients were accompanied by xanthomas and coronary artery disease. Elevated apo A-I levels were due to an increased synthetic rate, while the elevation of LDL-apo B resulted from both an enhanced synthetic rate and a prolonged residence time. Another case was also identified, who showed similar kinetic patterns of apo A-I and apo B. How-ever, the molecular defect of these cases is unknown. Table 1
Etiology of primary and secondary hyperalphalipoproteinemia (HALP)
Primary(familial)HALP CETP deficiency Familial HL deficiency
Familial HALP with premature corneal opacity and reduced HL activity
Familial HALP with reduced uptake of HDL by lymphocytes Familial HALP due to overproduction of apo A-I
Familial HALP associated with variant apo C-III (Lys58Glu)
Familial HALP and hyperbetalipoproteinemia
Secondary(non-familial)HALP Long-term massive intake of alcohol Primary biliary cirrhosis
HALP with albumin complexing Multiple lipomatosis
Emphysema Drugs:
Corticosteroids Insulin, sucrose/insulin Estrogen
Fibrates (clofibrate, bezafibrate, gemfibrozil, etc) Nicotinic acid and its derivatives
HMG-CoA reductase inhibitor Phenytoin
Kobayashi et al. reported a case of familial HALP whose lymphocytes showed a reduced uptake of HDL [26]. However, the molecular basis for HALP in this case has not been clarified. A polymorphism due to an A to G transition, 78 base pairs upstream from the transcription start site of human apo A-I gene, was shown to be associated with HALP in women, but not in men [27]. However, Minnich et al. demonstrated in French Canadians that this substitution did not directly confer high HDL-cholesterol levels, but may be in linkage disequilibrium with other sequence polymor-phism(s) at this locus in a subset of alleles that raise HDL-cholesterol levels [28]. Von Eckardstein et al. reported a family of HALP with variant apolipoprotein C-III (Lys58Glu) [29]. The patients were het-erozygous for the mutation and plasma apo C-III con-centrations were decreased by 30 – 40%. Large HDL particles enriched with apo E were observed in the patients, although the mechanism of HALP in this family is unknown.
2.2. Secondary HALP
HALP is also associated with a variety of diseases and factors. Alcohol consumption is accompanied by an increase in serum HDL-cholesterol [30]. However, in heavy alcohol drinkers the high HDL-cholesterol level can turn low when they suffer from liver cirrhosis. Alcohol consumption reduces plasma CETP activity and protein mass [31]. Hirano et al. reported similar findings in chronic heavy alcohol drinkers [32]. Small polydisperse LDL, which is one of the characteristic features of CETP deficiency, was observed in heavy alcohol drinkers with markedly reduced CETP activity. Furthermore, chronic alcohol intake induced changes in both plasma concentration and size of HDL. The size of HDL particles was large in alcohol drinkers. The cessation of alcohol was accompanied by a reduction in serum HDL-cholesterol with a concomitant decrease in HDL particle size. The LDL in chronic alcohol drinkers was poor in CE and rich in TG. These LDL particles had a lower affinity than normal LDL to the LDL receptors of human skin fibroblasts [33]. After cessation of alcohol, the affinity of LDL to LDL recep-tors was normalized. These changes were in parallel with those of CETP activity and mass, suggesting that alcohol intake may directly affect the plasma CETP level, thereby controlling the concentration and compo-sition of LDL and HDL.
Patients with primary biliary cirrhosis (PBC) are often associated with a marked HALP. The levels of serum total cholesterol, apo A-I, apo C-III and apo E are also elevated [34]. The elevation of HDL-cholesterol is attributed solely to the increase in HDL2-cholesterol. The HDL2 particles of the patients are enriched with apo E and larger in size than those of controls. HL
activity and mass are decreased in these patients, while CETP activity and mass are elevated. Reduced HL activity may partly contribute to the pathogenesis of HALP in PBC. In the PBC patients associated with apo E2/2 [35], serum HDL-cholesterol level was extremely high (244 and 263 mg/dl). In these patients, the LDL density fraction contained HDL1-like particles with apo A-I, and HDLc-like particles with predominant apo E and a trace amount of apo A-I. These HDLc-like particles were similar to those observed in CETP-defi-cient patients. Therefore, HL and apo E-mediated HDL removal by the liver may be important in the metabolism of HDL (Fig. 1).
An HALP patient with albumin complexing and elevated IgM level was reported [36]. The patient’s plasma contained a unique complex of albumin with apo A-I. HL was decreased and lipoprotein lipase was increased in this patient, which may result in an in-crease in HDL2 with an enrichment of phospholipids and thereby with an increased affinity for albumin present in the very high density lipoprotein (VHDL) fraction. HALP is also caused by corticosteroids, su-crose and insulin, estrogen, hypolipidemic drugs such as HMG-CoA reductase inhibitors and fibrates, phenytoin and chlorinated hydrocarbons (Table 1). Taken to-gether, HALP is a syndrome and the lipoprotein profile and its atherogenicity may differ between each type of HALP.
3. Serum HDL-cholesterol level does not correlate with anti-atherogenicity — implications from transgenic animal models
Patients with genetic HDL deficiency do not always present premature atherosclerosis. Subjects with apo
A-IMILANOare protected from atherosclerosis, although
the level of serum HDL-cholesterol is low [37]. Al-though this was not due to a relatively increased capac-ity of mutant apo A-IMILANO to recruit membrane cholesterol [38,39], an intravenous infusion of recombi-nant apo A-IMILANO/phospholipid complex to apo E-deficient mice fed a high-cholesterol diet from 20 until 25 weeks induced a reduction of aortic lipid and macrophage content and also inhibited an increase in aortic atherosclerosis [40].
Fig. 1. Metabolism of high density lipoprotein (HDL). CE, cholesteryl ester; CETP, cholesteryl ester transfer protein; FC, free cholesterol; HL, hepatic lipase; LCAT, lecithin:cholesterol acyltransferase; LPL, lipoprotein lipase; OxLDL, oxidized LDL; SRA, scavenger receptor class A type I and type II; SR-BI, scavenger receptor class B type I.
function than those from control subjects [42]. Further-more, recent data of transgenic animal models have provided an insight into the physiological function of various molecules involved in remodelling of HDL and their relationship with atherosclerosis.
3.1. Apo A-I and A-II transgenic mice
Transgenic mice overexpressing human apo A-I [43,44] showed an increase in serum HDL-cholesterol, human apo A-I and HDL particle size and were resis-tant to diet-induced atherosclerosis [45]. However, in apo A-I knockout mice, in which apo A-I was totally deficient and serum HDL-cholesterol was decreased by 80% [46], atherosclerosis was not observed even after feeding an atherogenic diet [47].
In contrast to apo A-I overexpressing mice, trans-genic mice overexpressing mouse apo A-II showed ac-celerated atherosclerosis despite increased serum HDL-cholesterol levels [48,49]. Human apo A-I and apo A-I/A-II transgenic mice were developed in an atherosclerosis-susceptible strain. After feeding an atherogenic diet, the area of atherosclerotic lesions in the apo A-I/A-II transgenic mice was 15-fold greater than in the apo A-I transgenic mice despite similar total cholesterol and HDL-cholesterol levels. Thus, the protein composition of HDL may significantly affect its role in atherogenesis and HDL with apo A-I is more antiatherogenic than HDL with apo A-I/A-II.
3.2. LCAT transgenic mice and rabbits
In transgenic mice overexpressing human LCAT [50], serum total cholesterol and HDL-cholesterol were in-creased, HDL was enlarged, and enhanced atheroscle-rosis was observed after feeding an atherogenic diet compared to control mice [51]. The composition and function of LCAT transgenic mouse HDLs were abnor-mal, and the uptake by the liver of [3H]CE incorporated in HDL was reduced by 41%. These data suggested an ineffective transport of HDL-cholesterol to the liver in LCAT transgenic mice. The transgenic rabbits overex-pressing human LCAT were also hyperalphalipo-proteinemic and the size of HDL was enlarged similar to LCAT transgenic mice, however these rabbits were resistant to atherosclerosis [52,53]. In the LCAT knock-out mice [54], a tendency of reduced atherosclerosis was observed, although the difference was not statistically significant [55]. These discrepancies may derive from species difference and most possibly from the fact that mice are deficient in CETP activity. The lipoprotein metabolism of mice is distinctively different from that of humans.
3.3. HL transgenic mice and rabbits
in HL transgenic mice. Transgenic rabbits overexpress-ing HL also showed a marked reduction of plasma HDL and IDL [57], but atherosclerotic lesions in the ascending aorta and the aortic arch were thicker in the transgenic rabbits than in the non-transgenic rabbits [58].
3.4. CETP transgenic mouse models
Plasma CETP is a hydrophobic glycoprotein with a Mr of 70 – 74 kDa [1,2]. Human CETP consists of 476 amino acid residues. Human CETP gene is located in chromosome 16 near the LCAT gene and consists of 16 exons and 15 introns [59]. CETP is synthesized by the liver, spleen, adipose tissues, small intestines, adrenal gland, kidney, heart and skeletal muscles, and by cells including monocyte-derived macrophages, B-lymphocytes, and adipocytes. CETP activity is very low in mice and rats and high in rabbits. In CETP trans-genic mice with mouse metallothionein-I promoter [60], the transgene overexpression after zinc induction was detected in the liver and small intestines, which caused a decrease in serum cholesterol, HDL-cholesterol and HDL particle size. The changes in lipoprotein profile were more striking in transgenic mice expressing cynomolgus monkey CETP [61,62], than in those devel-oped by Agellon [60]. In the human CETP transgenic mice [60], plasma HDL-cholesterol levels of zinc-treated male and female transgenic mice fell by 27 and 15%, respectively, on a chow diet. In contrast, in the simian CETP transgenic mice [61,62], plasma HDL-cholesterol levels fell by 50%. CETP transgenic mice with a CETP minigene linked to the natural flanking se-quences of human CETP gene showed a 2.5-fold in-crease in plasma CETP activity in response to a high-fat, high-cholesterol diet [63]. The CETP trans-genic mice mated with hypertriglyceridemic human apo C-III transgenic mice showed a marked decrease in HDL-cholesterol and apo A-I and in the size of HDL, which was due to an increased fractional catabolic rate of HDL-CE and apo A-I [64]. Hayek et al have shown that CETP overexpression inhibited the development of early atherosclerotic lesions in hypertriglyceridemic apo C-III transgenic mice [65]. As described above, human LCAT transgenic mice lead to increased plasma HDL-cholesterol levels, but paradoxically enhanced atherosclerosis. LCAT transgenic mice were crossbred with CETP transgenic mice [66]. The expression of CETP normalized both the plasma clearance of CE from HDL as well as the liver uptake of CE from HDL in LCAT transgenic mice. On the atherogenic diet, the mean aortic lesion area was reduced by 41% in LCAT/
CETP double transgenic mice compared to LCAT transgenic mice. Thus, CETP expression was considered to reduce atherosclerosis in LCAT transgenic mice by correcting the dysfunctional properties of
LCAT-trans-genic mouse HDL and promoting the hepatic uptake of HDL-CE.
3.5. SR-BI transgenic mouse models
The class B scavenger receptor, SR-BI, is an HDL receptor which is involved in the selective uptake of CE from HDL [3]. It is abundantly expressed in the steroidogenic tissues and liver. Adenovirus-mediated hepatic overexpression of SR-BI in mice on both sinu-soidal and canalicular surfaces of hepatocytes induced almost total disappearance of plasma HDL and a sub-stantial increase in biliary cholesterol [67]. This effect may be due either to increased hepatic HDL-CE uptake or to enhanced secretion of cholesterol into bile, or both. Liver specific overexpression of SR-BI decreased the levels of VLDL apo B, LDL apo B, and HDL-cholesterol [68]. To examine the expression of SR-BI on atherosclerosis, LDL receptor-deficient mice were fed a high-fat/high-cholesterol diet for 2 or 12 weeks to in-duce atherosclerotic lesions of different stages [69]. These mice were then injected with a recombinant adenovirus encoding murine SR-BI. Transient hepatic overexpression of SR-BI in mice with both early and advanced lesions decreased atherosclerosis, despite de-creased cholesterol levels. The average HDL-cholesterol levels were significantly correlated with the size of atherosclerotic lesions. Ueda et al. [70] devel-oped SR-BI transgenic mice, which showed lower plasma levels and accelerated clearance of HDL. Fur-thermore, they examined two lines of SR-BI transgenic mice with high (10-fold increases) and low (2-fold in-creases) in SR-BI expression in an inbred mouse back-ground hemizygous for a human apo B transgene [71]. The low expression SR-BI/apo B transgenics had more than a 2-fold decrease in the development of diet-in-duced fatty streak lesions compared to the apo B transgenics, however the high expression SR-BI/apo B transgenics with a marked decrease in HDL-cholesterol showed an atherogenic response similar to that of the apo B transgenics. Thus, the expression of SR-BI was considered to be antiatherogenic under the conditions of moderate SR-BI expression. Furthermore, decreased atherosclerosis has been shown in heterozygous LDL receptor knockout mice expressing SR-BI despite de-creased HDL-cholesterol [72]. Therefore, the uptake of CE by liver SR-BI may be a key step which determines the efficiency of reverse cholesterol transport system and possibly the development of atherosclerosis.
size as well as in controlling cholesterol concentrations in bile. Regarding the atherosclerosis in SR-BI knock-out mice, virtually no detectable lesions were observed at relatively young age (4 – 7 weeks), however a signifi-cant lesion developed in the SR-BI/apo E double knockout mice in the aortic root and coronary arteries [75]. Moreover, increased LDL cholesterol and atherosclerosis were demonstrated in LDL receptor-deficient mice with attenuated expression of scavenger receptor B1, which also showed a slight increase in HDL-cholesterol [76]. The effect of attenuated SR-BI expression was partly due to increased LDL cholesterol levels in this model. Taken together, the data on SR-BI overexpression and knockout mice suggest that upregu-lation of SR-BI could have therapeutic potential for the treatment of atherosclerosis despite low HDL-choles-terol levels. It is also suggested that reduced expression of hepatic SR-BI may be proatherogenic despite high HDL-cholesterol levels, since reverse cholesterol trans-port is impaired in this condition.
3.6. Pitfalls of transgenic animal models
These animal models have provided a very good tool for assessing the physiological roles of molecules. How-ever, there are difficulties in drawing conclusions on the atherogenicity of each molecule from studies using ani-mal models such as mice, of which the lipoprotein metabolism is quite different from that of humans. One has to assess the species differences in lipoprotein metabolism. Furthermore, these studies may establish a notion that serum levels of HDL-cholesterol do not necessarily correlate with the degree of protection from atherosclerosis. It may be crucial to assess the efficiency of reverse cholesterol transport system irrespective of serum HDL-cholesterol levels, since HDL particles be-come ‘dysfunctional’ due to the impairment of this system.
4. HALP due to plasma CETP deficiency
HALP patients due to plasma CETP deficiency have been reported mostly from Japan. There are some reports of CETP deficiency from German, Caucasian and Asian populations.
4.1. Gene defects in CETP deficiency
Concerning the gene defects in CETP deficiency, a G-to-A mutation in the 5%-splice donor site of intron 14 was initially identified in Japanese CETP-deficient sub-jects [77 – 79]. This mutation is common in the Japanese HALP subjects and in the general population (het-erozygotes: about 1 – 2%) [79,80]. A missense mutation (442D:G) in the exon 15 was later reported [81].
Al-though the patients were heterozygous for this defect, they had a 3-fold increase in plasma HDL-cholesterol and markedly decreased plasma CETP activity and mass, suggesting that the 442D:G mutation may have dominant effects on CETP and HDL in vivo. This was confirmed in vitro by expressing wild-type and mutant proteins in COS cells. The frequency of heterozygotes for the 442 D:G mutation is also high in the Japanese general population and HALP subjects [82,83].
A nonsense CETP mutation in exon 10 which causes premature stop codon was reported [84]. Another non-sense mutation in exon 6 (G181X) has recently been identified, which causes a total absence of CETP activ-ity [85]. This mutation is also common, although the frequency in HALP subjects is not so high compared with those of intron 14 splicing defect and 442 D:G mutation. Moreover, another defect in the intron 10 splice donor site of CETP gene has been identified, which causes exon 10 skipping, resulting in abnormal downstream splice site selection [86]. Although most of CETP-deficient subjects were found in the Asian popu-lation, novel mutations have recently been identified in HALP subjects from the German, Caucasian and Asian populations [87 – 90]. Fig. 2 summarizes the site of CETP gene mutations reported so far.
4.2. Abnormal plasma lipoproteins in CETP deficiency
Several procedures have been developed to determine CETP activity [91,92]. For example, the CE portion of donor lipoproteins (HDL3) is radiolabeled with [
14C] or
[3H]cholesterol, and unlabeled LDL or the dB1.063 g/ml fraction is used as a source of CETP followed by incubation at 37°C. After adding donor and acceptor lipoproteins to the assay buffer, a small amount of plasma or ad\1.21 g/ml plasma fraction is added as a source of CETP followed by incubation at 37°C. To further evaluate the CETP level, CETP mass assays are performed [93,94].
Fig. 2. Mutations in the human cholesteryl ester transfer protein (CETP) gene reported so far.
In native polyacrylamide gradient gel electrophoresis, the LDL particles in CETP deficiency are small and heterogeneous (polydisperse LDL) and HDL particles are extremely large [97]. By equilibrium density gradient ultracentrifugation, the LDL of CETP-deficient pa-tients comprises a group of heterogeneous lipoprotein particles distributed in a wide density range without a prominent peak, while the LDL from normal controls consists of a homogeneous group of lipoprotein parti-cles distributed in a narrower density range with a single sharp peak [99]. The LDL in each subfraction derived from patients is poor in CE and enriched with TG. In native polyacrylamide gradient gel electrophore-sis, each subfraction of control LDL contains only one species of homogeneous LDL particles that decreases in size with an increase in the density. In contrast, each subfraction of patients’ LDL contains two species of LDL particles; smaller LDL particles are found in addition to those which are identical to the normal control LDL particles observed in the corresponding subfractions. The IDL of the patients is also composed of two species of lipoproteins. Thus, two metabolic pathways may exist in the process of mature LDL formation [99]. VLDL is secreted by the liver as two species of lipoprotein particles different in size, and each type of VLDL is successively metabolized to LDL through IDL by a separate pathway. Various modula-tions might be involved in producing cholesterol-rich LDL, which possesses a high affinity for LDL recep-tors. The hydrolysis of TG by lipoprotein lipase and HL is crucial for this process. Furthermore, CETP plays an important role in converting small TG-rich LDL particles to large CE-rich homogeneous LDL particles by transferring CE from HDL.
As mentioned earlier, HALP is caused by various genetic or acquired abnormalities in lipoprotein
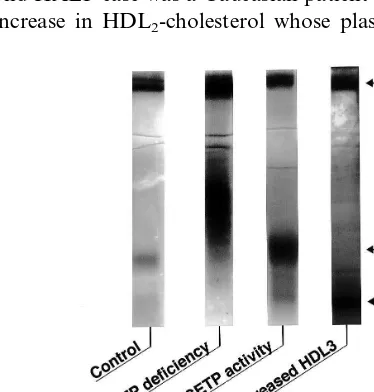
metabolism. The differences in lipoprotein profiles are remarkable between CETP deficiency and the other types of HALP. Fig. 3 demonstrates a native polyacry-lamide gradient gel electrophoretic pattern of the three HALP cases. One was a homozygous CETP deficiency whose HDL particles were extremely large (HDLc or HDL1) and LDL particles were polydisperse; the sec-ond HALP case was a Caucasian patient with a marked increase in HDL2-cholesterol whose plasma CETP
ac-Fig. 3. Comparison of a native polyacrylamide gradient gel elec-trophoretic pattern of hyperalphalipoproteinemic cases. In cholesteryl ester transfer protein (CETP) deficiency, very large high density lipoprotein (HDL) particles (HDL1or HDLc) as well as polydisperse
low density lipoprotein (LDL) particles are observed. In the hyperal-phalipoproteinemia (HALP) case with normal CETP activity, HDL2
particles are predominant. In contrast, in a peculiar HALP case with normal CETP activity, HDL3particles are predominant. The HALP
tivity was normal; the third case was also a Caucasian HALP subject whose CETP activity was normal and the increase in HDL-cholesterol was attributed solely to the increment of HDL3-cholesterol. The LDL of the latter two patients was monodisperse and the size of HDL particles was much smaller than that of CETP deficiency (HDL2size in the second case and HDL3size in the third case, respectively). It can be speculated that the HALP of the third case may result from an over-production of apo A-I. This type of HALP may be accompanied by an increased number of small HDL particles and could be anti-atherogenic.
4.3. Impaired function of lipoprotein particles in CETP-deficiency
The HDL2 particles from normal controls can pro-tect macrophages from cholesterol accumulation when incubated with acetylated LDL, and enhance choles-terol efflux from cells. In contrast, CE-rich large HDL2 particles from CETP-deficient subjects have a reduced capacity for inhibiting acetyl LDL-induced accumula-tion of CE in mouse peritoneal macrophages when HDL together with acetylated LDL is added to the culture medium, although the patients’ HDL3 particles show normal preventive effect against cholesterol accu-mulation [100]. Moreover, the HDL2from the patients has a less capacity than normal HDL2 for cholesterol efflux from macrophages preloaded with CE by acety-lated LDL. These data suggest that large CE-rich HDL2 particles in CETP deficiency do not have anti-atherogenic functions. Two types of HDL particles exist in human plasma; HDL particles which contain only apo A-I (designated as LpA-I) and those which contain both apo A-I and apo A-II (LpA-I/A-II). LpA-I level is decreased in CETP deficiency, resulting in a significant decrease in the efflux and LCAT-mediated esterification of cholesterol compared to normal con-trols [101].
Stable isotope study demonstrated that the fractional catabolic rates (FCRs) of apo A-I and A-II were signifi-cantly reduced in the CETP-deficient patients compared with the controls, while the production rates of apo A-I and apo A-II were normal [102]. Therefore, ho-mozygous CETP deficiency causes a markedly delayed catabolism of apo A-I and apo A-II. Regarding the metabolism of apo B, the FCRs of apo B were signifi-cantly increased in the patients, while the production rates of apo B were slightly decreased [103]. Moreover, the apo B-containing TG-rich LDL particles from CETP-deficient patients had a lower affinity to LDL receptor than those from controls [104]. These LDL particles may be susceptible to oxidation in vivo. Taken together, CETP deficiency causes functional abnormali-ties as well as compositional changes of both HDL and LDL.
The effect of CETP on the lipoproteins of CETP-deficient patients was evaluated in vitro [105]. The addition of CETP into the patients’ plasma induced a disappearance of large HDL particles and a formation of VHDL-like particles. The addition of HL accelerated this process. The VHDL-like particles isolated by gel permeation chromatography were enriched with phos-pholipids and protein. The protein moiety was almost solely apo A-I. These particles migrated at the preb -po-sition and partly at the a-position and showed a more increased capacity than normal HDL3 for cholesterol efflux from acetyl LDL-loaded mouse peritoneal macrophages. This finding is supported by the recent report of Fancone et al, showing increased preb-HDL levels, cholesterol efflux and LCAT-mediated esterifica-tion in vivo in mice overexpressing human CETP and human apo A-I transgenes [106]. Furthermore, HDL particles of human CETP transgenic mice had a more efficient capability of cholesterol efflux than those of non-transgenic mice, and the co-expression of both human apo A-I and CETP improved the efficiency of HDL particles in the human apo A-I/CETP transgenic mice when compared with the human apo A-I trans-genic mice [107]. Thus, CETP may be essential for remodeling of large HDL particles into small ones with a potent anti-atherogenic function.
5. Is HALP anti-atherogenic or proatherogenic?
5.1. Possible enhancement of atherosclerosis in CETP deficiency
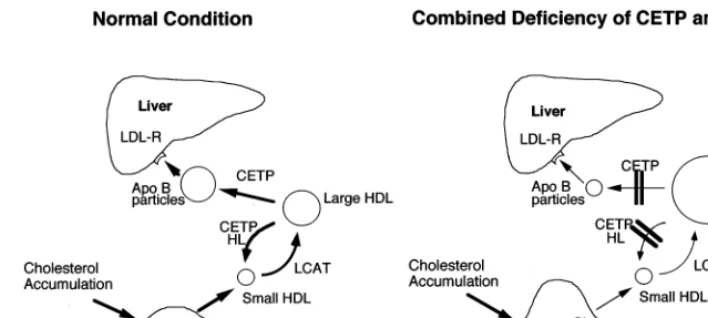
Kuiven-Fig. 4. Metabolic abnormalities in hyperalphalipoproteinemia (HALP) associated with a combined reduction of cholesteryl ester transfer protein (CETP) and HTGL (Ref. [113]).
hoven et al recently observed a significant dose-depen-dent association of Taq B1B2 genotype marker with the progression of atherosclerosis in the placebo group, while this association was abolished by pravastatin-treatment [112]. Pravastatin therapy slowed the pro-gression of coronary atherosclerosis in B1B1 carriers (with higher CETP levels), but not in B2B2 carriers (with lower CETP levels). This common DNA variant appeared to predict whether men with coronary artery disease will benefit from treatment with pravastatin to delay the progression of coronary atherosclerosis.
However, corneal arcus was noted in CETP-deficient cases [113] and a relatively high prevalence of cere-brovascular diseases was observed in certain CETP-deficient families. Some of the homozygous and heterozygous subjects with CETP deficiency were ac-companied by atherosclerotic cardiovascular diseases such as coronary heart disease [113,114], cerebrovascu-lar disease and arteriosclerosis obliterans [113]. In the HALP subjects whose CETP and HL activities were both reduced, atherosclerotic cardiovascular diseases occurred [113]. The metabolic abnormalities in HALP associated with a combined reduction of CETP and HL is illustrated in Fig. 4. The increased HDL2 particles in CETP-deficient subjects have a reduced capacity for cholesterol efflux [100]. CETP together with HL trans-forms large HDL particles into small ones, which have a potent antiatherogenic function [105]. Collet et al. reported that the HDL isolated from mice expressing CETP showed a 2 – 4-fold increase in SR-BI-mediated HDL-CE uptake compared to HDL from mice lacking CETP [115]. The addition of CETP to HDL in cell culture did not lead to increased selective uptake of HDL-CE by cells. However, when human HDL was enriched with TG by incubation with TG-rich lipo-proteins in the presence of CETP, then treated with HL, a significant enhancement of HDL-CE uptake was observed. Therefore, the remodelling of human HDL
by CETP, involving CE-TG interchange, followed by the action of HL, leads to the enhanced uptake of HDL-CE by SR-BI. As mentioned earlier, SR-BI knockout mice show a marked increase in HDL-choles-terol and HDL size, however atherosclerosis is acceler-ated in atherosclerosis-susceptible mice. The enlarged HDL particles in CETP deficiency resemble those ob-served in SR-BI knockout mice and these HDL parti-cles in vivo may be ‘dysfunctional’.
whose HDL-cholesterol level was between 41 and 60 mg/dl [117]. However, men with increased HDL levels (\60 mg/dl) had a low risk of coronary artery disease irrespective of the CETP genotype. Moriyama et al reported no significant difference in the prevalence of coronary heart disease between HALP subjects with and without CETP mutations [118]. They showed that HALP subjects (HDL-cholesterol\80 mg/dl as well as HDL-cholesterol from 60 to 79 mg/dl) appeared to be protected against coronary heart disease irrespective of the presence or absence of CETP deficiency. The reason for the discrepancy between the data of Omagari and Kochi remains to be clarified. The D442G mutation which has a less remarkable effect on CETP activity than the intron 14 splicing defect was predominant in this Kochi population, while the intron 14 splicing defect with a stronger effect on CETP activity was markedly frequent in Omagari area. These differences in the genotype of CETP deficiency might be consid-ered when such a discrepancy is evaluated.
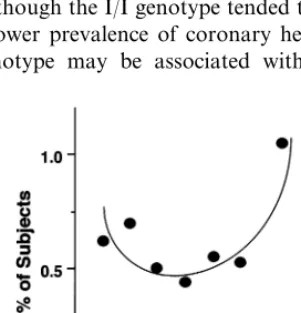
Furthermore, a CETP polymorphism (I405V) was studied in the Honolulu Heart Program cohort to de-termine the relationship between the polymorphism, CETP and HDL levels, and coronary heart disease [119]. The V/V genotype was associated with lower CETP concentrations than the I/V or I/I genotype. The HDL-cholesterol levels were higher in men with the V/V genotype than in men with the I/V or I/I genotype. However, the increase in HDL was only significant in hypertriglyceridemic men with V/V genotype. Although the prevalence of coronary heart disease was not signifi-cantly different among the three genotypes, it was significantly higher among V/V than I/V or I/I subjects. Thus, although the I/I genotype tended to be associated with a lower prevalence of coronary heart disease, the V/V genotype may be associated with higher
HDL-cholesterol levels and increased coronary heart disease in hypertriglyceridemic men. As reported recently by Agerholm-Larsen et al., women not treated with hor-mone replacement therapy, who were heterozygous or homozygous for Val405 and were associated with lower CETP activity and higher HDL-cholesterol level, had a 1.4 – 2.1-fold increase in the risk of ischemic heart dis-ease, although no significant associations were detected in men [120]. Thus, it was speculated that CETP defi-ciency may not be anti-atherogenic, but rather proatherogenic in humans. However, a careful prospec-tive study on the atherogenicity of patients with CETP deficiency is necessary to draw conclusions. It is also essential to determine clearly whether the increase in HDL-cholesterol may protect against or rather acceler-ate atherosclerosis in CETP-deficient patients.
5.2. Atherogenicity of other types of HALP
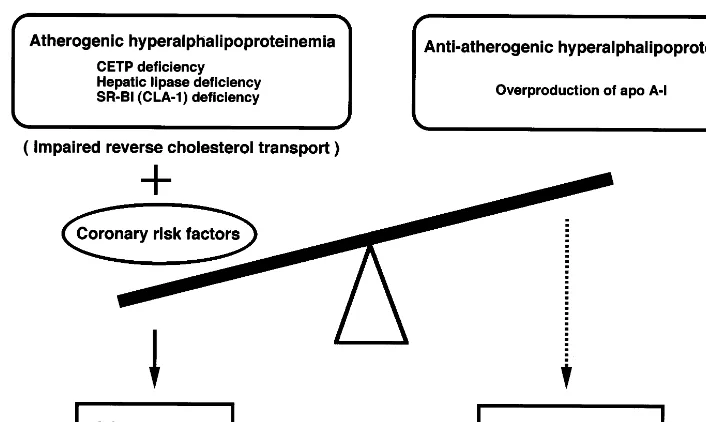
The atherogenicity of other types of HALP may depend upon the etiology of HALP. For example, HALP due to HL deficiency may be proatherogenic, while that due to overproduction of apo A-I could be anti-atherogenic (such as HALP with predominant in-crease in HDL3 as demonstrated in Fig. 3). Fig. 6 illustrates the determinants of atherogenicity in HALP subjects. Its relation to atherogenicity may depend upon the presence or absence of other coronary risk factors and also upon the efficiency of reverse choles-terol transport system. Furthermore, the number of preb-HDL particles in tissues may determine the atherogenicity, since these particles were shown to be anti-atherogenic and produced by CETP [101], HL [108] and phospholipid transfer protein (PLTP) [111]. Physicians should carefully follow up HALP patients with dysfunctional HDL due to impaired reverse cholesterol transport system to avoid the development of atherosclerosis.
In this review, the current knowledge on the patho-genesis of HALP was presented with special reference to the lipoprotein and gene abnormalities and athero-genicity of CETP deficiency. CETP plays a crucial role in the reverse cholesterol transport system. Patients with CETP deficiency are sometimes accompanied by atherosclerosis and thus CETP deficiency could rather be designated as ‘atherogenic HALP’. To the authors’ current knowledge, the level of serum HDL may not necessarily imply the functional aspects of HDL parti-cles. HALP may be a condition of an impaired reverse cholesterol transport system. The ‘dysfunctional HDL particles’ produced by the impairment of molecules involved in reverse cholesterol transport system may lead to the acceleration of atherosclerosis in humans. For this reason, CETP inhibitors may not be a good tool for the purpose of treatment of atherosclerosis. Taken together, it may be important to establish a Fig. 5. Relationship between plasma high density lipoprotein
(HDL)-cholesterol levels and the incidence of ischemic ECG changes in Omagari (modified from Ref. [116]). , denote the incidence of ST-segment depression of \1 mm in the subjects with each
Fig. 6. Classification of hyperalphalipoproteinemia (HALP) in terms of atherogenicity.
strategy to assess the efficiency of reverse cholesterol transport system rather than merely determining the level of HDL-cholesterol.
Acknowledgements
This work was partly supported by a Grant-in-Aid from the Japanese Ministry of Education, Science, Sports and Culture (No. 04404085, 05670589, 07671135). The authors thank Takeshi Arai and Masato Ishigami for their great contribution to CETP-related research.
References
[1] Tall A. Lipid transfer proteins. Annu Rev Biochem 1995;64:235 – 57.
[2] Lagrost L. Regulation of cholesteryl ester transfer protein (CETP) activity; review of in vitro and in vivo studies. Biochem Biophys Acta 1994;1215:209 – 36.
[3] Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996;271:518 – 20.
[4] Glomset JA. The plasma lecithin:cholesterol acyltransferase reaction. J Lipid Res 1968;9:155 – 67.
[5] Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res 1995;36:211 – 28.
[6] Hill SA, McQueen MJ. Reverse cholesterol transport — a review of the process and its clinical implications. Clin Biochem 1997;30:517 – 25.
[7] Castro GR, Fielding CJ. Early incorporation of cell-derived cholesterol into pre-beta-migrating high-density lipoprotein. Biochemistry 1988;27:25 – 9.
[8] Barrans A, Jaspard B, Barbaras R, Chap H, Perret B, Collet X. Pre-beta HDL: structure and metabolism. Biochem Biophys Acta 1996;1300:73 – 85.
[9] Rothblat GH, de la Llera-Moya M, Atger V, Kellner-Weibel G, Williams DL, Phillips MC. Cell cholesterol efflux: integration of old and new observations provides new insights. J Lipid Res 1999;40:781 – 96.
[10] Fidge NH. High density lipoprotein receptors, binding proteins, and ligands. J Lipid Res 1999;40:187 – 201.
[11] Ji Y, Jian B, Wang N, Sun Y, de la Llera Moya M, Phillips MC, Rothblat GH, Swaney JB, Tall AR. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem 1997;272:20982 – 5.
[12] Hirano K, Yamashita S, Nakagawa Y, Ohya T, Matsuura F, Tsukamoto K, Okamoto Y, Matsuyama A, Matsumoto K, Miyagawa J, Matsuzawa Y. Expression of human scavenger receptor class B type I in cultured human monocyte-derived macrophages and atherosclerotic lesions. Circ Res 1999;85:108 – 16.
[13] Brooks-Wilson A, Marcil M, Clee SM, Zhang L-H, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HOF, Loubser O, Ouelette BFF, Fichter K, Ashbourne-Excoffon KJD, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J Jr, Hayden MR. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet 1999;22:336 – 45.
[14] Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcu-rumez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet 1999;22:347 – 51.
[15] Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, Assmann G. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet 1999;22:352 – 5. [16] Glueck CJ, Gartside P, Fallat RW, Sielski J, Steiner PM. Longevity syndromes; familial hypobeta and familial hyperal-phalipoproteinemia. J Lab Clin Med 1976;88:941 – 57. [17] Matsuzawa Y, Yamashita S, Kameda K, Kubo M, Tarui S,
[18] Matsuzawa Y, Yamashita S, Funahashi T, Yamamoto A, Tarui S. Selective reduction of cholesterol in HDL2 fraction by probucol in familial hypercholesterolemia and hyper HDL2 cholesterolemia with abnormal cholesteryl ester transfer. Am J Cardiol 1988;62:66B – 72B.
[19] Groener JEM, Da Col PG, Kostner GM. A hyperalphalipo-proteinemic family with normal cholesteryl ester transfer/ ex-change activity. Biochem J 1987;242:27 – 32.
[20] Patsch W, Kuisk I, Glueck C, Schonfeld G. Lipoproteins in familial hyperalphalipoproteinemia. Arteriosclerosis 1981;1:156 – 61.
[21] Hegele RA, Little JA, Vezina C, Maguire GF, Tu L, Wolever TS, Jenkins DJ, Connelly PW. Hepatic lipase deficiency. Clini-cal, biochemiClini-cal, and molecular genetic characteristics. Arte-rioscler Thromb 1993;13:720 – 8.
[22] Brand K, Dugi KA, Brunzell JD, Nevin DN, Santamarina-Fojo S. A novel AG mutation in intron I of the hepatic
lipase gene leads to alternative splicing resulting in enzyme deficiency. J Lipid Res 1996;37:1213 – 23.
[23] Breckenridge WC, Little JA, Alaupovic P, Wang CS, Kuksis A, Kakis G, Lindgren F, Gardiner G. Lipoprotein abnormalities associated with a familial deficiency of hepatic lipase. Atherosclerosis 1982;45:161 – 79.
[24] Rader DJ, Schaefer JR, Lohse P, Ikewaki K, Thomas F, Harris WA, Zech LA, Dujovne CA, Brewer HB Jr. Increased produc-tion of apolipoprotein A-I associated with elevated plasma levels of high-density lipoproteins, apolipoprotein A-I, and lipoprotein A-I in a patient with familial hyperalphalipo-proteinemia. Metabolism 1993;42:1429 – 34.
[25] Schmidt HH, Gregg RE, Tietge UJ, Beisiegel U, Zech LA, Brewer HB Jr, Manns MP, Bojanovski D. Upregulated synthe-sis of both apolipoprotein A-I and apolipoprotein B in familial hyperalphalipoproteinemia and hyperbetalipoproteinemia. Metabolism 1998;47:1160 – 6.
[26] Kobayashi J, Nishide T, Shinomiya M, Sasaki N, Shirai K, Saito Y, Yoshida S. A familial hyperalphalipoproteinemia with low uptake of high density lipoproteins into peripheral lymphocytes. Atherosclerosis 1988;73:105 – 11.
[27] Pagani F, Sidoli A, Giudici GA, Barenghi L, Vergani C, Baralle FE. Human apolipoprotein A-I gene promoter polymorphism; association with hyperalphalipoproteinemia. J Lipid Res 1990;31:1371 – 7.
[28] Minnich A, DeLangavant G, Lavigne J, Roederer G, Lussier-Cacan S, Davignon J. GA substitution at position-75 of the
apolipoprotein A-I gene promoter. Evidence against a direct effect on HDL cholesterol levels. Arterioscler Thromb Vasc Biol 1995;15:1740 – 5.
[29] von Eckardstein A, Holz H, Sandkamp M, Weng W, Funke H, Assmann G. Apolipoprotein C-III (Lys58Glu). Identification
of an apolipoprotein C-III variant in a family with hyperal-phalipoproteinemia. J Clin Invest 1991;87:1724 – 31.
[30] Giziano JM, Buring JE, Breslow JL, Goldhaber SZ, Rosner B, VanDenburgh M, Willett W, Hennekens CH. Moderate alcohol intake, increased levels of high-density lipoprotein and its sub-fractions, and decreased risk of myocardial infaction. N Engl J Med 1993;329:1829 – 34.
[31] Hannuksela M, Marcel YL, Kesaniemi YA, Savolainen MJ. Reduction in the concentration and activity of plasma cholesteryl ester transfer protein by alcohol. J Lipid Res 1992;33:737 – 44.
[32] Hirano K, Matsuzawa Y, Sakai N, Hiraoka H, Nozaki S, Funahashi T, Yamashita S, Kubo M, Tarui S. Polydisperse low density lipoproteins in hyperalphalipoproteinemic chronic alco-hol drinkers in association with marked reduction of calco-holesteryl ester transfer protein activity. Metabolism 1992;41:1313 – 8. [33] Hirano K, Yamashita S, Sakai N, Hiraoka H, Ueyama Y,
Funahashi T, Matsuzawa Y. Low-density lipoproteins in
hyper-alphalipoproteinemic heavy alcohol drinkers have reduced affinity for the low-density lipoprotein receptor. Clin Biochem 1992;25:357 – 62.
[34] Hiraoka H, Yamashita S, Matsuzawa Y, Kubo M, Nozaki S, Sakai N, Hirano K, Kawata S, Tarui S. Decrease of hepatic triglyceride lipase and increase of cholesteryl ester transfer protein in patients with primary biliary cirrhosis-Relation to the abnormalities in high density lipoprotein. Hepatology 1993;18:103 – 10.
[35] Hiraoka H. Impairment of the reverse cholesterol transport system associated with primary biliary cirrhosis. Med J Osaka Univ 1991;43:399 – 408.
[36] Demacker PN, Jansen RT, Hijmans AG, van Gorp HP. A case of hyperalphalipoproteinemia associated with albumin com-plexing. Atherosclerosis 1994;111:13 – 23.
[37] Breslow JL. Familial disorders of high-density lipoprotein metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Dis-ease. 7th ed New York: McGraw-Hill, 1995: 2031 – 52. [38] Franceschini G, Calabresi L, Chiesa G, Parolini C, Sirtori CR,
Canavesi M, Bernini F. Increased cholesterol efflux potential of sera from apo A-IMilano carriers and transgenic mice.
Arte-rioscler Thromb Vasc Biol 1999;19:1257 – 62.
[39] Calabresi L, Canavesi M, Bernini F, Franceschini G. Cell cholesterol efflux to reconstituted high-density lipoproteins con-taining the apolipoprotein A-IMilano dimer. Biochemistry
1999;38:16307 – 14.
[40] Shah PK, Nilsson J, Kaul S, Fishbein MC, Ageland H, Ham-sten A, Johansson J, Karpe F, Cercek B. Effects of recombi-nant apolipoprotein A-I (Milano) on aortic atherosclerosis in apolipoprotein E-deficient mice. Circulation 1998;97:780 – 5. [41] Franceschini G, Sirtori M, Vaccarino V, Gianfranceschi G,
Rezzonico L, Chiesa G, Sirtori CR. Mechanisms of HDL reduction after probucol. Changes in HDL subfractions and increased reverse cholesteryl ester transfer. Arteriosclerosis 1989;9:462 – 9.
[42] Ishigami M, Yamashita S, Sakai N, Hirano KI, Arai T, Maruyama T, Takami S, Koyama M, Kameda-Takemura K, Matsuzawa Y. High-density lipoproteins from probucol-treated patients have increased capacity to promote cholesterol efflux from mouse peritoneal macrophages loaded with acetylated low-density lipoproteins. Eur J Clin Invest 1997;27:285 – 92. [43] Walsh A, Ito Y, Breslow JL. High levels of human
apolipo-protein A-I in transgenic mice result in increased plasma levels of small high density lipoprotein (HDL) particles comparable to human HDL3. J Biol Chem 1989;264:6488 – 94.
[44] Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size classes. Proc Natl Acad Sci USA 1991;88:434 – 8.
[45] Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein A-I. Nature 1991;353:265 – 7.
[46] Williamson R, Lee D, Hagaman J, Maeda N. Marked reduc-tion of high density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc Natl Acad Sci USA 1992;89:7134 – 8.
[47] Li H, Reddick RL, Maeda N. Lack of apo A-I is not associated with increased susceptibility to atherosclerosis in mice. Arte-rioscler Thromb 1993;13:1814 – 21.
[48] Schultz JR, Verstuyft JG, Gong EL, Nichols AV, Rubin EM. Protein composition determines the anti-atherogenic properties of HDL in trangenic mice. Nature 1993;365:761 – 4.
[50] Vaisman BL, Klein HG, Rouis M, Berard AM, Kindt MR, Talley GD, Meyn SM, Hoyt RF Jr, Marcovina SM, Albers JJ, Hoeg JM, Brewer HB Jr, Santamarina-Fojo S. Overexpression of human lecithin cholesterol acyltransferase leads to hyperal-phalipoproteinemia in transgenic mice. J Biol Chem 1995;270:12269 – 75.
[51] Berard AM, Foger B, Remaley A, Shamburek R, Vaisman BL, Talley G, Paigen B, Hoyt RF Jr, Marcovina S, Brewer HB Jr, Santamarina-Fojo S. High plasma HDL concentrations associ-ated with enhanced atherosclerosis in transgenic mice overex-pressing lecithin-cholesteryl acyltransferase. Nat Med 1997;3:744 – 9.
[52] Brousseau ME, Santamaria-Fojo S, Zech LA, Berard AM, Vaisman BL, Meyn SM, Powell D, Brewer HB Jr, Hoeg JM. Hyperalphalipoproteinemia in human lecithin cholesterol acyl-transferase transgenic rabbits. In vivo apolipoprotein A-I catabolism is delayed in a gene dose-dependent manner. J Clin Invest 1996;97:1844 – 51.
[53] Hoeg JM, Santamarina-Fojo S, Berard AM, Cornhill JF, Herd-erick EE, Feldman SH, Haudenschild CC, Vaisman BL, Hoyt RF Jr, Demosky SJ Jr, Kauffman RD, Hazel CM, Marcovina SM, Brewer HB Jr. Overexpression of lecithin:cholesterol acyl-transferase in transgenic rabbits prevents diet-induced atherosclerosis. Proc Natl Acad Sci USA 1996;93:11448 – 53. [54] Sakai N, Vaisman BL, Koch CA, Hoyt RF Jr, Meyn SM,
Talley GD, Paiz JA, Brewer HB Jr, Santamarina-Fojo S. Targeted disruption of the mouse lecithin:cholesterol acyltrans-ferase (LCAT) gene. Generation of a new animal model for human LCAT deficiency. J Biol Chem 1997;272:7506 – 10. [55] Sakai N, Vaisman BL, Koch CA, Paiz JA, Brewer HB Jr,
Santamarina-Fojo S, Paigen B. Analysis of aortic atherosclero-sis and glomeruloscleroatherosclero-sis in LCAT knockout mice. Circulation 1997;96:I-229.
[56] Busch SJ, Barnhart RL, Martin GA, Fitzgerald MC, Yates MT, Mao SJ, Thomas CE, Jackson RL. Human hepatic triglyc-eride lipase expression reduces high density lipoprotein and aortic cholesterol in cholesterol-fed transgenic mice. J Biol Chem 1994;269:16376 – 82.
[57] Fan J, Wang J, Bensadoun A, Lauer SJ, Dang Q, Mahley RW, Taylor JM. Overexpression of hepatic lipase in transgenic rab-bits leads to a marked reduction of plasma high density lipo-proteins and intermediate density lipolipo-proteins. Proc Natl Acad Sci USA 1994;91:8724 – 8.
[58] Taylor JM, Fan J. Transgenic rabbit models for the study of atherosclerosis. Frontiers Biosci 1997;2:298 – 308.
[59] Agellon L, Quinet E, Gillette T, Drayna D, Brown M, Tall AR. Organization of the human cholesteryl ester transfer protein gene. Biochemistry 1990;29:1372 – 6.
[60] Agellon LB, Walsh A, Hayek T, Moulin P, Jiang XC, Shelanski SA, Breslow JL, Tall AR. Reduced high density lipoprotein cholesterol in human cholesteryl ester transfer protein trans-genic mice. J Biol Chem 1991;266:10796 – 801.
[61] Marotti KR, Castle CK, Murray RW, Rehberg EF, Polites HG, Melchior GW. The role of cholesteryl ester transfer protein in primate apolipoprotein A-I metabolism; insights from studies with transgenic mice. Arterioscler Thromb 1992;12:736 – 44.
[62] Marotti KR, Castle CK, Boyle TP, Lin AH, Murray RW, Melchior GW. Severe atherosclerosis in transgenic mice ex-pressing simian cholesteryl ester transfer protein. Nature 1993;364:73 – 5.
[63] Jiang XC, Agellon LB, Walsh A, Breslow JL, Tall A. Dietary cholesterol increases transcription of the human cholesteryl ester transfer protein gene in transgenic mice; dependence on natural flanking sequences. J Clin Invest 1992;90:1290 – 5. [64] Hayek T, Azrolan N, Verdery RB, Walsh A, Chajek-Shaul T,
Agellon LB, Tall AR, Breslow JL. Hypertriglyceridemia and
cholesteryl ester transfer protein interact to dramatically alter high density lipoprotein levels, particle sizes, and metabolism; studies in transgenic mice. J Clin Invest 1993;92:1143 – 52. [65] Hayek T, Masucci-Magoulas L, Jiang X, Walsh A, Rubin E,
Breslow JL, Tall AR. Decreased early atherosclerotic lesions in hypertriglyceridemic mice expressing cholesteryl ester transfer protein transgene. J Clin Invest 1995;96:2071 – 4.
[66] Foger B, Chase M, Amar MJ, Vaisman BL, Shamburek RD, Paigen B, Fruchart-Najib J, Paiz JA, Koch CA, Hoyt RF, Brewer HB Jr, Santamarina-Fojo S. Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin cholesterolacyltrans-ferase transgenic mice. J Biol Chem 1999;274:36912 – 20. [67] Kozarsky KF, Donahee MH, Rigotti A, Iqbal SN, Edelman
ER, Krieger M. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature 1997;387:414 – 7.
[68] Wang N, Arai T, Ji Y, Rinninger F, Tall AR. Liver-specific overexpression of scavenger receptor BI decreases levels of very low density lipoprotein apo B, low density lipoprotein apo B, and high density lipoprotein in transgenic mice. J Biol Chem 1998;273:32920 – 6.
[69] Kozarsky KF, Donahee MH, Glick JM, Krieger M, Rader DJ. Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL recep-tor-deficient mouse. Arterioscler Thromb Vasc Biol 2000;20:721 – 7.
[70] Ueda Y, Royers L, Gong E, Zhang J, Cooper PN, Francone O, Rubin EM. Lower plasma levels and accelerated clearance of plasma high density lipoprotein (HDL) band non-HDL choles-terol in scavenger receptor class B type I transgenic mice. J Biol Chem 1999;274:7165 – 71.
[71] Ueda Y, Gong E, Royer L, Cooper PN, Francone OL, Rubin EM. Relationship between Expression Levels and Atherogene-sis in Scavenger Receptor Class B, Type I Transgenics. J Biol Chem 2000;275:20368 – 73.
[72] Arai T, Wang N, Bezouevski M, Welch C, Tall AR. Decreased atherosclerosis in heterozygous low density lipoprotein recep-tor-deficient mice expressing the scavenger receptor BI trans-gene. J Biol Chem 1999;274:2366 – 71.
[73] Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci USA 1997;94:12610 – 5.
[74] Varban ML, Rinninger F, Wang N, Fairchild-Huntress V, Dunmore JH, Fang Q, Gosselin ML, Dixon KL, Deeds JD, Acton SL, Tall AR, Huszar D. Targeted mutation reveals a central role for SR-BI in hepatic selective uptake of high density lipoprotein cholesterol. Proc Natl Acad Sci USA 1998;95:4619 – 24.
[75] Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci USA 1999;96:9322 – 7.
[76] Huszar D, Varban ML, Rinninger F, Feeley R, Arai T, Fairchild-Huntress V, Donovan MJ, Tall AR. Increased LDL cholesterol and atherosclerosis in LDL receptor-deficient mice with attenuated expression of scavenger receptor B1. Arte-rioscler Thromb Vasc Biol 2000;20:1068 – 73.
[78] Yamashita S, Hui DY, Wetterau JR, Sprecher DL, Harmony JAK, Matsuzawa Y, Tarui S. Total deficiency of plasma cholesteryl ester transfer protein in subjects homozygous and heterozygous for the intron 14 splicing defect. Biochem Biophys Res Commun 1990;170:1346 – 51.
[79] Inazu A, Brown ML, Hesler CB, Agellon LB, Koizumi J, Takata K, Maruyama Y, Mabuchi H, Tall AR. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med 1990;323:1234 – 8.
[80] Hirano K, Yamashita S, Funahashi T, Sakai N, Menju M, Ishigami M, Hiraoka H, Takemura K, Tokunaga K, Mat-suzawa Y. Frequency of intron 14 splicing defect of cholesteryl ester transfer protein gene in the Japanese general population. Atherosclerosis 1993;100:85 – 90.
[81] Takahashi K, Jiang X-C, Sakai N, Yamashita S, Hirano K, Bujo H, Yamazaki H, Kusunoki J, Miura T, Kussie P, Mat-suzawa Y, Saito Y, Tall A. A missense mutation in the cholesteryl ester transfer protein gene with dominant effects on plasma high density lipoproteins. J Clin Invest 1993;92:2063 – 4. [82] Sakai N, Yamashita S, Hirano K, Menju M, Arai T, Kobayashi K, Ishigami M, Yoshida Y, Hoshino T, Nakajima N, Kameda-Takemura K, Matsuzawa Y. Frequency of exon 15 missense mutation (442D:G) in cholesteryl ester transfer protein gene in hyperalphalipoproteinemic Japanese subjects. Atherosclerosis 1995;114:139 – 45.
[83] Inazu A, Jiang X, Haraki T, Yagi K, Kamon N, Koizumi J, Mabuchi H, Takeda R, Takata K, Moriyama Y, Doi M, Tall AR. Genetic cholesteryl ester transfer protein deficiency caused by two prevalent mutations as a major determinant of increased levels of high density lipoprotein cholesterol. J Clin Invest 1994;94:1872 – 82.
[84] Gotoda T, Kinoshita M, Shimano H, Harada K, Shimada M, Ohsuga J, Teramoto T, Yazaki Y, Yamada N. Cholesteryl ester transfer protein deficiency caused by a nonsense mutation detected in the patient’s macrophage mRNA. Biochem Biophys Res Commun 1993;194:519 – 24.
[85] Arai T, Yamashita S, Sakai N, Hirano K, Okada S, Ishigami M, Maruyama T, Yamane M, Kobayashi H, Nozaki S, Funa-hashi T, Kameda-Takemura K, Nakajima N, Matsuzawa Y. A novel nonsense mutation (G181X) in the human cholesteryl ester transfer protein gene in Japanese hyperalphalipoproteine-mic subjects. J Lipid Res 1996;37:2145 – 54.
[86] Sakai N, Santamarina-Fojo S, Yamashita S, Matsuzawa Y, Brewer HB Jr. Exon 10 skipping caused by intron 10 splice donor site mutation in cholesteryl ester transfer protein gene results in abnormal downstream splice site selection. J Lipid Res 1996;37:2065 – 73.
[87] Funke H, Wiebusch H, Fuer L, Muntoni S, Schulte H, Ass-mann G. Identification of mutations in the cholesteryl ester transfer protein in Europeans with elevated high density lipo-protein cholesterol. Circulation 1994;90(Part 2):241.
[88] Teh EM, Dolphin PJ, Breckenridge WC, Tan M-H. Human plasma CETP deficiency; identification of a novel mutation in exon 9 of the CETP gene in a Caucasian subject from North America. J Lipid Res 1998;39:442 – 56.
[89] Hill SA, Nazir DJ, Jayaratne P, Bamford KS, McQueen MJ. Mutations in cholesteryl ester transfer protein and hepatic lipase in a North American population. Clin Biochem 1997;30:413 – 8.
[90] Ritsch A, Drexel H, Amann FW, Pfeifhofer C, Patsch JR. Deficiency of cholesteryl ester transfer protein. Description of the molecular defect and the dissociation of cholesteryl ester and triglyceride transport in plasma. Arterioscler Thromb Vasc Biol 1997;17:3433 – 41.
[91] Tollefson JH, Albers JJ. Isolation, characterization, and assay of plasma lipid transfer proteins. Methods Enzymol 1986;129:797 – 816.
[92] Kato H, Nakanishi T, Arai H, Nishida HI, Nishida T. Purifica-tion, microheterogeneity, and stability of human lipid transfer protein. J Biol Chem 1989;264:4082 – 7.
[93] Marcel YL, McPherson R, Hogue M, Czarnecka H, Zawadzki Z, Weech PK, Whitlock ME, Tall AR, Milne RW. Distribution and concentration of cholesteryl ester transfer protein in plasma of normolipidemic subjects. J Clin Invest 1990;85:10 – 7. [94] Fukasawa M, Arai H, Inoue K. Establishment of anti-human
cholesteryl ester transfer protein monoclonal antibodies and radioimmunoassaying of the level of cholesteryl ester transfer protein in human plasma. J Biochem 1992;111:696 – 8. [95] Kurasawa T, Yokoyama S, Miyake Y, Yamaura T, Yamamoto
A. Deficiency of serum cholesteryl ester transfer between high and low density lipoproteins in human serum and a case with decreased transfer rate in association with hyperalphalipo-proteinemia. J Biochem 1985;98:1499 – 508.
[96] Koizumi J, Mabuchi H, Yoshimura A, Michishita I, Takeda M, Itoh H, Sakai Y, Sakai T, Ueda K, Takeda R. Deficiency of serum cholesteryl-ester transfer activity in patients with familial hyperalphalipoproteinemia. Atherosclerosis 1985;58:175 – 86. [97] Yamashita S, Matsuzawa Y, Okazaki M, Kako H, Yasugi T,
Akioka H, Hirano K, Tarui S. Small polydisperse low density lipoproteins in familial hyperalphalipoproteinemia with com-plete deficiency of cholesteryl ester transfer activity. Atheroscle-rosis 1988;70:7 – 12.
[98] Yamashita S, Sprecher DL, Sakai N, Matsuzawa Y, Tarui S, Hui DY. Accumulation of apolipoprotein E-rich high density lipoproteins in hyperalphalipoproteinemic human subjects with plasma cholesteryl ester transfer protein deficiency. J Clin In-vest 1990;86:688 – 95.
[99] Sakai N, Matsuzawa Y, Hirano K, Yamashita S, Nozaki S, Ueyama Y, Kubo M, Tarui S. Detection of two species of low density lipoprotein particles in cholesteryl ester transfer protein deficiency. Arterioscler Thromb 1991;11:71 – 9.
[100] Ishigami M, Yamashita S, Sakai N, Arai T, Hirano K, Hiraoka H, Takemura K, Matsuzawa Y. Large and cholesteryl ester-rich high density lipoproteins in cholesteryl ester transfer protein deficiency can not protect macrophages from cholesterol accu-mulation induced by acetylated low density lipoproteins. J Biochem 1994;116:257 – 62.
[101] Ohta T, Nakamura R, Takata K, Saito Y, Yamashita S, Horiuchi S, Matsuda I. Structural and functional differences of subspecies of apoA-I-containing lipoprotein in patients with plasma cholesteryl ester transfer protein deficiency. J Lipid Res 1995;36:696 – 704.
[102] Ikewaki K, Rader DJ, Sakamoto T, Nishiwaki M, Wakimoto N, Schaefer JR, Ishikawa T, Fairwell T, Zech LA, Nakamura H, Brewer HB Jr. Delayed catabolism of high density lipo-protein apolipolipo-proteins A-I and A-II in human cholesteryl ester transfer protein deficiency. J Clin Invest 1993;92:1650 – 8. [103] Ikewaki K, Nishiwaki M, Sakamoto T, Ishikawa T, Fairwell T,
Zech LA, Nagano M, Nakamura H, Brewer HB Jr, Rader DJ. Increased catabolic rate of low density lipoproteins in humans with cholesteryl ester transfer protein deficiency. J Clin Invest 1995;96:1573 – 81.
[104] Sakai N, Yamashita S, Hirano K, Ishigami M, Arai T, Kobayashi K, Funahashi T, Matsuzawa Y. Decreased affinity of low density lipoprotein (LDL) particles for LDL receptors in patients with cholesteryl ester transfer protein deficiency. Eur J Clin Invest 1995;25:332 – 9.
[105] Yamashita S, Ishigami M, Arai T, Sakai N, Hirano K, Kameda-Takemura K, Tokunaga K, Matsuzawa Y. Very high density lipoprotein (VHDL) induced by plasma cholesteryl ester transfer protein (CETP) have a potent anti-atherogenic func-tion. Ann N Y Acad Sci USA 1995;748:606 – 8.
esterifica-tion in mice expressing the human cholesteryl ester transfer protein (CETP) and human apolipoprotein A-I (apo A-I) trans-gene. J Lipid Res 1996;37:1268 – 77.
[107] Atger V, de la Llera Moya M, Bamberger M, Francone O, Cosgrove P, Tall A, Walsh A, Moatti N, Rothblat G. Choles-terol efflux potential of sera from mice expressing human cholesteryl ester transfer protein and/or human apolipoprotein A-I. J Clin Invest 1995;96:2613 – 22.
[108] Quinet E, Tall A, Ramakrishnan R, Rudel L. Plasma lipid transfer protein as a determinant of the atherogenicity of monkey plasma lipoproteins. J Clin Invest 1991;87:1559 – 66. [109] Sugano M, Makino N. Changes in plasma lipoprotein
choles-terol levels by antisense oligodeoxynucleotides against cholesteryl ester transfer protein in cholesterol-fed rabbits. J Biol Chem 1996;271:19080 – 3.
[110] Sugano M, Makino N, Sawada S, Otsuka S, Watanabe M, Okamoto H, Kamada M, Mizushima A. Effects of antisense oligodeoxynucleotides against cholesteryl ester transfer protein on the development of atherosclerosis in cholesterol-fed rabbits. J Biol Chem 1998;273:5033 – 6.
[111] Albers JJ, Tu A-Y, Wolfbauer G, Cheung MC, Marcovina SM. Molecular biology of phospholipid transfer protein. Curr Opin Lipidol 1996;7:88 – 93.
[112] Kuivenhoven JA, Jukema JW, Zwinderman AH, de Knijff P, McPherson R, Bruschke AVG, Lie KI, Kastelein JJP, for the Regression Growth Evaluation Statin Study Group. The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. N Engl J Med 1998;338:86 – 93.
[113] Hirano K, Yamashita S, Kuga Y, Sakai N, Nozaki S, Kihara S, Arai T, Yanagi K, Takami S, Menju M, Ishigami M, Yoshida Y, Kameda-Takemura K, Hayashi K, Matsuzawa Y. Atherosclerotic disease in marked hyperalphalipoproteinemia; Combined reduction of cholesteryl ester transfer protein and hepatic triglyceride lipase. Arterioscler Thromb Vasc Biol 1995;15:1849 – 56.
[114] Hirano K, Yamashita S, Sakai N, Arai T, Yoshida Y, Nozaki S, Kameda-Takemura K, Matsuzawa Y. Molecular defect and atherogenicity in cholesteryl ester transfer protein deficiency. Ann N Y Acad Sci 1995;748:606 – 8.
[115] Collet X, Tall AR, Serajuddin H, Guendouzi K, Royer L, Oliveira H, Barbaras R, Jiang X-C, Francone OL. Remodeling of HDL by CETP in vivo and by CETP and hepatic lipase in vitro results in enhanced uptake of HDL-CE by cells expressing scavenger receptor B-I. J Lipid Res 1999;40:1185 – 93. [116] Hirano K, Yamashita S, Nakajima N, Arai T, Maruyama T,
Yoshida Y, Ishigami M, Sakai N, Kameda-Takemura K, Mat-suzawa Y. Genetic cholesteryl ester transfer protein deficiency is extremely frequent in the Omagari area of Japan; marked hyperalphalipoproteinemia caused by CETP gene mutation is not associated with longevity. Arterioscler Thromb Vasc Biol 1997;17:1053 – 9.
[117] Zhong S, Sharp DS, Grove JS, Bruce C, Yano K, Curb JD, Tall AR. Increased coronary heart disease in Japanese – Ameri-can men with mutations in the cholesteryl ester transfer protein gene despite increased HDL levels. J Clin Invest 1996;97:2917 – 23.
[118] Moriyama Y, Okamura T, Inazu A, Doi M, Iso H, Mouri Y, Ishikawa Y, Suzuki H, Iida M, Koizumi J, Mabuchi H, Ko-machi Y. A low prevalence of coronary heart disease among subjects with increased high-density lipoprotein cholesterol lev-els, including those with plasma cholesteryl ester transfer protein deficiency. Prev Med 1998;27:659 – 67.
[119] Bruce C, Sharp DS, Tall AR. Relationship of HDL and coronary heart disease to a common amino acid polymorphism in the cholesteryl ester transfer protein in men with and without hypertriglyceridemia. J Lipid Res 1998;39:1071 – 8.
[120] Agerholm-Larsen B, Nordestgaard BG, Steffensen R, Jensen G, Tybjaerg-Hansen A. Elevated HDL cholesterol is a risk factor for ischemic heart disease in white women when caused by a common mutation in the cholesteryl ester transfer protein gene. Circulation 2000;101:1907 – 12.