Reversible surface-sorption-induced electron-transfer oxidation
of Fe(II) at reactive sites on a synthetic clay mineral
A. Ge´hin
a, J.-M. Grene`che
b, C. Tournassat
c,*, J. Brendle´
d, D.G. Rancourt
e, L. Charlet
aaLGIT, Universite´ Grenoble-1, F 38041 Grenoble, France bLPEC, Universite´ du Maine, F 72085 Le Mans, France
cBRGM, Environment and Process Division (EPI/MIS), 3 avenue Claude Guillemin, F 45060 Orle´ans, France dLMPC, ENSC, F 68093 Mulhouse, France
eDepartment of Physics, University of Ottawa, Ottawa, Ont., Canada K1N 6N5
Received 6 April 2006; accepted in revised form 23 October 2006
Abstract
The sorption of57Fe(II) onto an Fe-free, mineralogically pure and Ca-saturated synthetic montmorillonite sample (structural formu-la: Ca0.15(Al1.4Mg0.6)(Si4)O10(OH,F)2), was studied as a function of pH under strictly anoxic conditions (N2glove box atmosphere, O2 content <1 ppm), using wet chemistry and cryogenic (T= 77 K)57Fe Mo¨ssbauer spectrometry. No Fe(III) was detected in solution at any pH. However, in pH conditions where Fe(II) is removed from solution, a significant amount of surface-bound Fe(III) was produced, which increased with pH from 0% to 3% of total Fe in a pre-sorption edge region (i.e. at pH < 7.5 where about 15% of total Fe is sorbed) to 7% of total Fe when all Fe is sorbed. At low pH, where the pre-sorption edge plateau occurs (2 < pH < 7.5), the total sorbed-Fe amount remained constant but, within this sorbed-Fe pool, the Fe(III)/Fe(II) ratio increased with pH, from 0.14 at pH 2 up to 0.74 at pH 7. The pre-sorption edge plateau is interpreted as cation exchange on interlayer surfaces together with a sorption phenomenon occurring on highly reactive (i.e. high affinity) surface sites. As pH increases and protons are removed from the clay edge surface, we propose that more and more of these highly reactive sites acquire a steric configuration that stabilizes Fe(III) relative to Fe(II), thereby inducing a Fe to clay particle electron transfer. A sorption model based on cation exchange combined with surface complexation and electron transfers reproduces both wet chemical as well as the Mo¨ssbauer spectrometric results. The mechanism is fully reversible: sorbed-Fe is reduced as pH decreases (Mo¨ssbauer solid-state analyses) and all Fe returned to solution is returned as Fe(II) (solution analyses). This would not be the case if the observed oxidations were due to contaminant oxidizing agents in solution. The present work shows that alternating pH may induce surface redox phenomena in the absence of an electron acceptor in solution other than H2O. 2006 Elsevier Inc. All rights reserved.
1. Introduction
Aqueous ferrous iron is a major cation and an impor-tant reducimpor-tant in a variety of natural anoxic environments (eutrophic lakes, ocean and swamp sediments, hydromor-phic soils, anoxic groundwaters and ocean basins), as well as in engineered systems, i.e. zero-valent iron permeable reactive barriers, or PRBs, and radioactive waste reposito-ry sites (e.g. Wehrli, 1990; Klausen et al., 1995; Charlet
et al., 1998a,b, 2002; Hofstetter et al., 1999, 2002, 2006; Pe-cher et al., 2002; Strathmann and Stone, 2003and referenc-es therein). At equilibrium, dissolved Fe(II) is controlled by siderite (FeCO3(s)), green rusts and Fe-rich calcite in sul-fide-poor, carbonate-rich surficial environments, and by FeSx(where1 <x< 2; pyrite forx= 2) in sulfide-rich
envi-ronments (Berner, 1971). In the former case, Fe(II) concen-trations range from 104 to 102.5M, depending on pH and PCO2(Emerson, 1976; Postma, 1982; Criaud and Fou-illac, 1986a,b), while in the latter case, Fe(II) concentra-tions are often in the 104–103M range (Emerson et al.,
1980; Balzer, 1982; Davison et al., 1999; Bott, 2002). In low PCO2, low PH2S environments, Fe(II) solubility is
0016-7037/$ - see front matter 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.gca.2006.10.019
* Corresponding author. Fax: +33 2 38 64 30 62. E-mail address:[email protected](C. Tournassat).
controlled by the precipitation of Fe(OH)2(s), Fe3O4 (mag-netite), green rust, or Fe3PO4(s). In granular, zero-valent iron PRBs and their outflows, Fe(II) concentrations typi-cally lie between 106 and 3·104 M (Naftz et al.,
2000). However, many surfacic systems are not at equilib-rium and are subject to cyclic redox fluctuations ( Pere-tyazhko and Sposito, 2005), e.g. as a result of seasonal climatic variations (Thompson et al., 2006). The migration of Fe(II) ions in aqueous environments is, as with most divalent cations, retarded by sorption on minerals with a large surface area and specific sorption properties such as clay minerals (Drever, 1971; Liger et al., 1999).
The clay sorption capacity has been well established for a wide range of potential pollutants in Na+and Ca2+ back-ground ionic media (e.g.Fletcher and Sposito, 1989; Char-let et al., 1993; Zachara et al., 1993; Zachara and Smith, 1994; Baeyens and Bradbury, 1997; Bradbury and Baeyens, 1997, 1999, 2002; Akcay, 1998; Turner et al., 1998). How-ever, the influence of other background ionic media such as Fe(II)-containing water is poorly documented. Previous studies of Fe(II) sorption processes on montmorillonite have shown that Fe(II) can sorb onto clay minerals in cat-ion exchange positcat-ion with a similar clay affinity for Fe(II) and Ca(II) (Kamei et al., 1999; Charlet and Tournassat, 2005). In the present study, new chemical and spectromet-ric experiments are proposed to characterize the mecha-nism of Fe(II) specific sorption on clay minerals.
We investigated the specific sorption of Fe(II) on a syn-thetic Fe-free clay mineral analogue of montmorillonite. The starting solid synthetic sorbant was devoid of any Fe, which is otherwise present in natural samples, either within the clay mineral or as accessory trace minerals. All the sorption experiments were carried out in strict anoxic conditions designed to prevent oxidation and to keep iron as ferrous iron. Solution chemistry and Mo¨ssbauer spec-trometry were used concomitantly. Results were analyzed in terms of (i) surface complexation and (ii) steric-driven reversible Fe oxidation in which electron transfer occurs between the sorbed-Fe centre and highly reactive clay edge functional groups.
2. Materials and methods
2.1. Chemicals
All solutions and suspensions were prepared with boiled, argon-degassed Millipore Milli-Q 18 MX water. NaOH and HCl stock solutions were made from Titrisol ampoules. The CaCl2and FeCl2stock solutions were pre-pared from analytical grade salts using the method de-scribed in Charlet and Tournassat (2005). The latter involves the dissolution of 100 mg of57Fe(0) in concentrat-ed HCl (0.1 mol l1) at100C. Then, the dissolved iron was placed in the glove box and diluted in deionized water to a final volume of 100 ml, to obtain a1000 ppm57Fe(II) stock solution. The very acidic conditions prevented the
oxidation of the suspension prior to its transfer in the glove box.
2.2. Clay material preparation
The synthesis of montmorillonite having the theoretical formula: Na0.30[(Al1.70Mg0.30)Si4O10(OH,F)2], nH2O was performed in acidic and fluoride medium under hydrother-mal conditions (Reinholdt et al., 2001). A hydrogel having a SiO2:0.212 Al2O3:0.075 MgO:0.075 Na2O:0.05 HF:96 H2O molar composition was prepared starting from water, hydrofluoric acid, sodium and magnesium acetate, silica and aluminum oxide. This mixture was matured for 2 h at room temperature before being hydrothermally treated in a PTFE lined stainless steel autoclave at 493 K for 72 h under autogenous water pressure. After crystalliza-tion, the product was separated by filtration and thorough-ly washed with distilled water. The pH of the supernatant was 3.5–4.5. After drying for one night at 338 K, the sam-ple was placed in a controlled humidity chamber (P/
P0= 80%, wherePrepresents the saturated vapor pressure over the sample at 298 K andP0the saturated aqueous va-por pressure at 298 K). Ca-montmorillonite was obtained by saturation of the prepared Na-montmorillonite with a 0.05 M aqueous solution of CaCl2. After treatment, the sol-id suspension was argon-degassed, before being placed in the glove box.
2.3. Clay material characterization
For XRD analysis, suspension aliquots were poured through a Millipore filter (0.4lm) and the clay cake was then deposited on a glass slide. The resulting oriented prep-arations were then dried at room temperature. XRD pat-terns were then recorded using a Bruker D5000 diffractometer (CuKa1,2 radiation: k= 0.15418 nm). The X-ray diffraction patterns of the synthetic Ca-montmoril-lonite (Fig. 1, grey line) show only basal 00‘ reflections, due to the oriented preparation of the samples. It compares well with a Ca-exchanged natural montmorillonite (MX80
Fig. 1. X-ray diffraction patterns of the Ca0.3[(Al1.4Mg0.6)Si4O10(OH,F)2]
synthetic montmorillonite (grey diffractogram) and the Ca0.14 (Al1.61
Mg0.24FeIII0:15Fe II
0:02) (Si3.98Al0.02) O10(OH)2MX80 montmorillonite (black
sample,Fig. 1, solid line). Thed001value is the same for the synthetic and the natural Ca-montmorillonite and equals 15.27 A˚ . The reflections close to 4.45–4.48 A˚ (020, 110) and 2.52–2.58 A˚ (130, 200) correspond to hk0 bands of dioctahedral smectites.
The cation exchange capacity of the synthetic montmo-rillonite were measured with the Cs–Li method described byAnderson and Sposito (1991)at pH 7. At this pH, the permanent structural charge (Cs) was found to be 0.44 eq/kg and the variable charge (Li) 0.19 eq/kg, accounting for a total CEC of 0.63 eq/kg at pH 7.
2.4. Fe(II) sorption and desorption experiments
Sorption experiments of Fe(II) on the synthetic mont-morillonite were followed by both solution chemistry and Mo¨ssbauer spectroscopy. The chemistry experiments were conducted in a N2 atmosphere glove box (Jacomex) in which the oxygen partial pressure (pO2) was monitored continuously with a Jacomex O2 sensor. The O2 content never exceeded 1 ppm in the glove box atmosphere, corre-sponding to a maximum solute O2 concentration of 0.13lmol/L.
In these experiments, the specific sorption of Fe(II) was studied as a function of pH on a short reaction time scale. Cation exchange between Fe(II) ions and the clay surface was minimized by using a 0.05 M CaCl2ionic background, which limits cation exchange by mass effect and maintains a constant total normality of the suspension. Experiments were carried out in closed 350 ml glass reactors with three inputs, one for the pH electrode and the two others for stock solution/suspension addition and sample extraction, respectively. An aliquot of clay stock suspension was added to CaCl2ionic background solution samples. The suspen-sion was acidified with HCl to pH 2 overnight (approxi-mately 16 h), upon which 650lM 57Fe(II) stock solution aliquot was added and the sorption experiments were started. Isotopically pure 57Fe(II) (> 97%) was used in order to enhance the Mo¨ssbauer signal. The sorption experiments proceeded by increasing the pH incrementally with successive additions of NaOH until all 57Fe(II) was sorbed. After each addition of NaOH, the suspension was allowed to equilibrate. Once the pH drift was less than 0.02 pH unit/10 min, a 10 ml sample of suspension was fil-tered through a 0.22lm pore size membrane and analyzed for iron concentration by induced coupled plasma atomic emission spectrometry (ICP-AES). A blank sample without montmorillonite was also prepared using the same experi-mental procedure with 10 mM ferrous chloride in a CaCl2 0.05 M ionic background. The Ferrozine method (Viollier et al., 2000) was also used to measure separately Fe(II) and Fe(III) concentrations in solutions up to 30lM after dilution and to check that no oxidation of Fe(II) to Fe(III) occurred in solution.
The desorption experiment was performed with the same procedure using HCl instead of NaOH and using the suspension obtained at the end of the sorption
experi-ment. As a control of the sorption reproducibility, the sus-pension was titrated once again with NaOH after the desorption experiment.
2.5.57Fe Mo¨ssbauer spectrometry
Mo¨ssbauer spectrometry was performed on a solid frac-tion obtained from these sorpfrac-tion experiments at five differ-ent pH values during the sorption experimdiffer-ent and one pH value during the desorption experiment. At these pH val-ues, once the pseudo-equilibrium was reached, small ali-quots (a few mL) of suspension were filtered (Millipore filter 0.02lm) and deposited on Mo¨ssbauer sample holders (diameter 1.5 cm). The sample holders were capped and then sealed with an epoxy resin. They were stored in the glove box before being taken out, immediately frozen in li-quid nitrogen and transported in a Dewar flask filled with liquid N2to the Mo¨ssbauer exchange-gas-bath cryostat.
The Mo¨ssbauer spectra were recorded at 77 K using a constant acceleration spectrometer and a57Co source dif-fusing into a rhodium matrix. Velocity calibrations were carried out using a-Fe foil at room temperature (RT, 295 K). All isomer shifts are reported relative to the a-Fe spectrum obtained at RT.
Two fitting models were considered to describe the Mo¨ssbauer spectra (which show a quadrupolar structure). The first one (MOSFIT: Teillet and Varret unpublished program) consists in using a discrete number of indepen-dent quadrupolar doublets of Lorentzian lines where the line width at half-height C (mm s1), the centre shift d (mm s1) and the quadrupole splittingDEQ(mm s1) were refined using a least-squared fitting procedure. The second modeling approach consists in using the Voigt-based meth-od of Rancourt and Ping (1991) for quadrupole splitting distributions (QSDs) with linear coupling to slave centre shift distributions (Rancourt, 1994a,b; Rancourt et al., 1994; Evans et al., 2005) as implemented in the commercial software Recoil(www.isapps.ca/recoil).
3. Results
3.1. Experimental verification of the anoxic conditions required for the study of Fe(II) sorption on montmorillonite
the preparation and the transport of the samples were car-ried out in the most strict anoxic conditions that can be achieved in a laboratory. Thus, no Fe(II) was accidentally oxidized by atmospheric O2, within the detection limits of our measurement methods.
3.2. Solution analyses
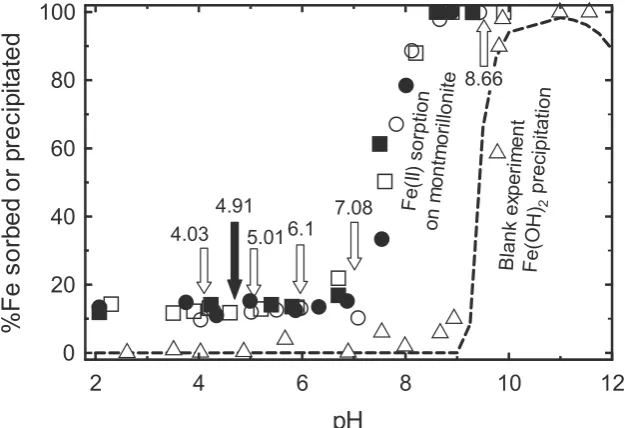
Fig. 3shows the results of iron solution analyses during the adsorption experiments. Fe(II) sorption results ob-tained with the Ferrozine method (Viollier et al., 2000) compare well with the values obtained by ICP-AES mea-surements. The Ferrozin method results indicated a Fe(III) concentration below the detection limit. This means that
100% of the iron in solution was in the ferrous oxidation state from pH 2 to 8.5. For pH > 8.5, more than 99% of the total iron was sorbed and the solute iron concentration was below the detection limit (1lM). In conclusion, the Ferrozin method analysis demonstrates that the totality of iron in solution was in the Fe(II) form, whatever the pH and the sorption events of the sample.
Without montmorillonite, the aqueous ferrous iron dis-appears from solution at a pH value about 9. The position of this edge is in full agreement with the precipitation of an amorphous ferrous iron hydroxide –Fe(OH)2– as shown by the calculated solubility curve inFig. 3. This result, togeth-er with the Fe(OH)2aging test, is good evidence of the well maintained anoxic conditions during the experiments. For the experiment with synthetic montmorillonite, a ‘‘sorption edge’’ was observed at a pH of about 7, preceded at lower pH by a sorption plateau. The Fe(II) and Fe(III) concen-trations in solution were also measured during increasing or decreasing pH cycles (between 2 and 9). The reproduc-ibility of the data demonstrates the total reversreproduc-ibility of the sorption phenomena. Therefore, a thermodynamics equilibrium approach can be used with confidence to de-scribe the sorption phenomena.
3.3.57Fe Mo¨ssbauer spectrometry
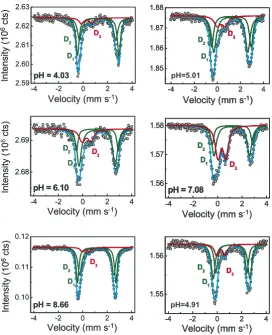
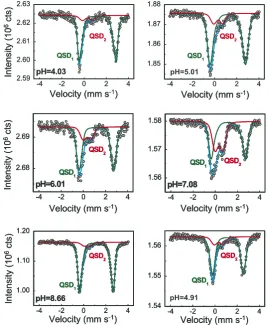
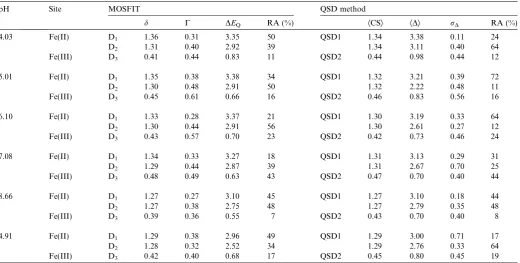
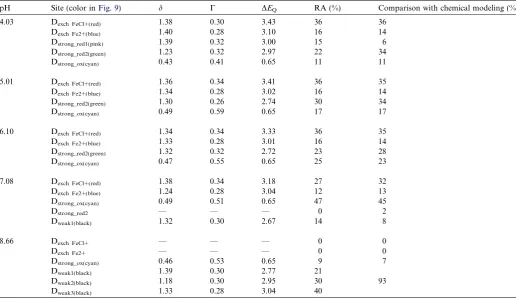
The paramagnetic Mo¨ssbauer spectra are shown inFigs. 4 and 5together with the hyperfine parameters (Table 1) obtained with the MOSFIT (Fig. 4) and the QSD modeling approach (Fig. 5), assuming two Gaussian QSD
Fig. 2. Mo¨ssbauer spectrum of ferrous hydroxide Fe(OH)2 at 77 K.
Hyperfine parameters:d= 1.29 mm s1, the isomer shift with respect to
metallica-Fe(0) at room temperature;DEQ= 3.09 mm s1, the
quadru-polar splitting;C= 0.32 mm s1, the full width at half height.
Fig. 3. Sorption edge, as a function of pH, of Fe(II) sorbed on synthetic montmorillonite ([SM] = 10 gclayL1, [Fe(II)]t= 630lM) in a 0.05 M CaCl2
background (open circles: first increasing pH sorption edge, total Fe measured by ICP-AES; closed circles: first decreasing pH sorption edge, total Fe measured by ICP-AES; open squares: second increasing pH sorption edge, Fe(II) measured by the Ferrozin method; closed squares: second decreasing pH sorption edge, Fe(II) measured by the Ferrozine method). The blank experiment results (without montmorillonite) are shown for comparison (open triangles: 100lM of Fe(II) in a 0.05 M CaCl2background). The dashed line is the computation of the Fe(OH)2solubility curve in the condition of the
components in a ferrous QSD and a single Gaussian QSD component for the ferric QSD. According to the values of center shift, the D1and D2doublets are clearly attributed to Fe(II) species while the D3doublet is assigned to Fe(III) species. Both models allow us to obtain unambiguously the Fe(III)/(Fe(II) + Fe(III)) ratio, i.e. the Fe(III) percentage of the total iron present in the solid sample, assuming equal Mo¨ssbauer recoilless fractions for ferric and ferrous iron. It is also important to emphasize that the values of some line widths appear to be larger than those expected for a highly crystalline structure with no chemical disorder.
The values of the hyperfine parameters do not corre-spond a priori to Fe present in any expected solid phases (e.g. ferrous hydroxide). During the pre-sorption edge (Fig. 3; pH 4.01, 5.01, 6.10 and 7.08), the Fe(III) compo-nent area increases with pH up to 43–44% of total-sorbed-Fe at pH 7.08. At the end of the main adsorption edge, it goes down to 7–8% of total-sorbed-Fe. When the pH returns to 4.91, the Mo¨ssbauer spectrum is very similar to the one observed before at pH 5.01 with the same rela-tive area of the total Fe(II) and Fe(III) components. How-ever, the isomer shifts and quadrupole splitting value are significantly different. Because the initial aqueous solu-tion containing iron is filtered after reacsolu-tion, the only
contribution to the Mo¨ssbauer spectra originates from the solid phase. Thus, we conclude that the hyperfine struc-ture is related to adsorbed iron.
The presence of Fe(III) signals in the Mo¨ssbauer spectra is unexpected. It cannot be explained by an accidental oxi-dation given all the care that was taken to avoid an acci-dental oxidation and, above all, the complete reversibility of the oxidation processes. Synthetic montmorillonite, as a material, is not likely to be considered as the oxidation agent since its elemental constituents Si(IV), Al(III) and Mg(II) are not redox reactive in our experimental conditions.
The comprehension of this surface chemistry is compli-cated by the fact that the exact structure of the surface of synthetic montmorillonite is not known. There are proba-bly a high number of different types of sorption sites in-volved in Fe(II) sorption, as for natural montmorillonite (Tournassat et al., 2004). This can be intuited by looking thoroughly at the hyperfine parameters given by the MOS-FIT approach. One notes that the Fe(II) quadrupole split-ting and centre shift values evolve with pH, providing evidence of changes in the sorbed-Fe(II) structural environ-ment. These changes can be due either to structural chang-es due to protonation/deprotonation of the sorbed iron or
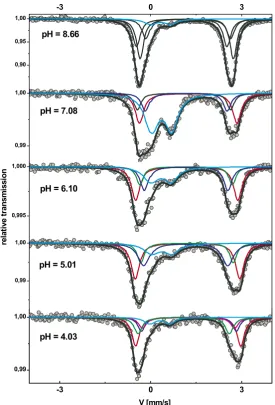
Fig. 4. Mo¨ssbauer spectra atT= 77 K shown together with hyperfine contributions obtained with the MOSFIT modeling approach (Table 1). On each spectrum, the pH of the suspensions is given. The paramagnetic doublets D1and D2with a large quadrupole splitting are characteristic of57Fe(II) surface
to a change in site population occupancy or both of these effects. Moreover, the pH 4.91 (return) and pH 5.01 (rais-ing) spectra are significantly different, despite arising in samples that have the same Fe(II) and sorbed-Fe(III) contents. This result provides strong evidence that the mechanism responsible for Fe(II) uptake and partial oxidation is due to several sites acting at different pH.
Mo¨ssbauer spectra were also fitted using the method of
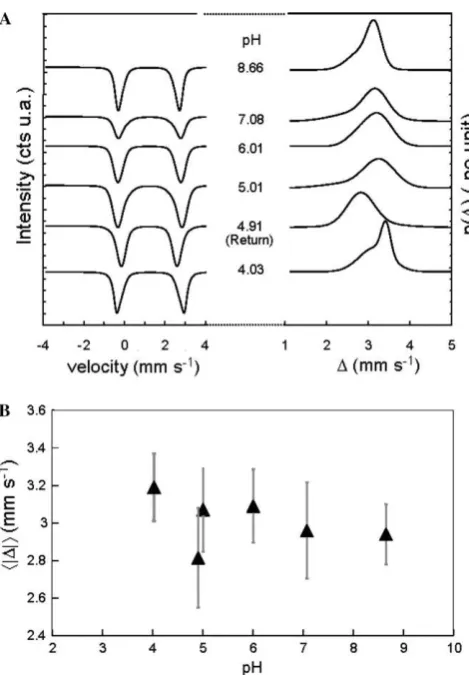
Rancourt and Ping (1991)based on QSDs in order to try to attribute the spectral changes in terms of local distortion of sorbed-Fe(II) environments (Rancourt, 1994a,b; Rancourt et al., 1994; Evans et al., 2005). We next discuss the Fe(II) QSDs, keeping in mind that each QSD characterizes a dis-tribution of sorbed-Fe(II) local environments (Rancourt, 1994a, 1998) on a given montmorillonite sample at a given pH. A Fe(II) QSD, therefore, characterizes the distribution of montmorillonite surface sites that accommodate Fe(II), including possible inner sphere coordinating anions on the solution side. Fig. 6A shows the Fe(II) QSDs extracted from the T= 77 K spectra of samples corresponding to the specific pH values depicted inFig. 3.
The sorbed-Fe(II) extracted QSDs allow the following main observations (Fig. 6): (1) The starting pH 4.03 Fe(II) QSD is significantly different from all the other Fe(II) QSD curves, being weighted more strongly towards larger QS
values and having a more narrow width. (2) The next incre-ments in pH, namely pH 5.01, 6.10 and 7.08 QSDs are very similar to each other and gradually evolve by narrowing and shifting to larger QS average values as pH increases. (3) The last increment in pH, namely pH 8.66, shows a QS shift towards low values together with a more narrow width. (4) As stated previously with the MOSFIT ap-proach, the pH 4.91 (return) and pH 5.01 (raising) QSD are significantly different, despite arising in samples that have the same sorbed-Fe(II) and sorbed-Fe(III) contents. Such characteristics are correlated to the hyperfine results obtained using the MOSFIT description.
The first Fe(II) binding sites (i.e., that binds at low pH) can be attributed mainly to cation exchanged Fe(II) in the interlayer (Fe2+and FeCl+). However, the signal cannot be attributed entirely to this species given its difference with previously published hyperfine parameters for exchanged Fe(II) at lower pH (Charlet and Tournassat, 2005). Other sites that bind Fe at low pH are then needed and are the so-called ‘‘high energy’’ sites in the following chemical modeling approach, those demonstrating the highest affin-ity towards Fe(II). The non-reversible QSD change in going from pH 5.01 to pH 8.66 (raising) appears to be pre-dominantly due to pH-induced changes in the Fe(II) local environments, probably including surface annealing
Fig. 5. Mo¨ssbauer spectra atT= 77 K shown together with hyperfine contributions obtained with the RECOIL modeling approach (Table 1). The quadrupole splitting distribution QSD1 with a large quadrupole splitting is characteristic of57Fe(II) surface species while the quadrupole splitting
around the newly complexed Fe(II). The proposed struc-tural annealing would produce the non-reversibility in local environment populations. The gradual changes in QSD on increasing pH through 6.01–8.66 are probably due to the combined effects of more and more low-affinity sites being occupied and pH-induced structural annealing.
3.4. Discussion on the oxidation mechanism and modeling approach
We have established that, under strict anoxic conditions, a charge transfer must occur so that some sorbed Fe(II) ends up being oxidized into sorbed Fe(III) atoms. The amount of observed Fe(III) exceeds by far the detected or expected limits of any chemical oxidants: O2 concentra-tion can not exceed 0.13lmol/L in the suspension whereas at least 7lmol/L Fe(II) was found to be oxidized into Fe(III) (sample at pH 4). Furthermore, the observed elec-tron transfer is reversible. This reversibility implies that the phenomenon is not due to an accidental contamination by oxidant. This is corroborated by a Mo¨ssbauer blank test carried out using the same handling procedure, but with a highly reactive synthetic ferrous hydroxide instead of the Fe(II)-montmorillonite system. In this test, no Fe(III) was detected (Fig. 2).
At first sight, such electron transfer is surprising. Ran-court et al. (2005) have recently described a mechanism
of sorbed-Fe redox stabilization, in the case of sorption-in-duced reduction of Fe(III) bacterial cell wall complex where the electron is transferred from the host cell to the sorbed-Fe(III). The same basic concept applies here for sorption-induced Fe(II) oxidation. In both cases, the differ-ence in Fe(II) versus Fe(III) complexation (or coordination or steric or ligand field stabilization) energy is involved and plays a dominant role. In both cases, the host is a high sur-face charge aqueous particle (bacterium or montmorillon-ite) with many different surface complexation sites, including a variety of multi-dentate sites.
The overall reaction could be described in terms of the following elemental steps. First, aqueous Fe(II) is sorbed at a specific surface site with high affinity for Fe2+as shown by the presence of specifically sorbed Fe(II) at pH 4 (see Mo¨ssbauer results):
FeðIIÞaqþsBðOHÞ3()sBðOHÞðO2Þ2Fe2þþ2Hþ
aq ð1Þ
where s„(OH)3represents not the montmorillonite solid, but the explicitly mentioned oxo or hydroxo coordinating surface functional group. Note the formal minus three charge of the reactive site does not preclude the overall charge of the site, as it is also controlled by the coordinated structural cations. Next, an electron transfer step is consid-ered, which oxidizes the sorbed Fe(II)
Table 1
Mo¨ssbauer hyperfine parameters of the spectra presented inFigs. 4 and 5
pH Site MOSFIT QSD method
d C DEQ RA (%) ÆCSæ ÆDæ rD RA (%)
4.03 Fe(II) D1 1.36 0.31 3.35 50 QSD1 1.34 3.38 0.11 24
D2 1.31 0.40 2.92 39 1.34 3.11 0.40 64
Fe(III) D3 0.41 0.44 0.83 11 QSD2 0.44 0.98 0.44 12
5.01 Fe(II) D1 1.35 0.38 3.38 34 QSD1 1.32 3.21 0.39 72
D2 1.30 0.48 2.91 50 1.32 2.22 0.48 11
Fe(III) D3 0.45 0.61 0.66 16 QSD2 0.46 0.83 0.56 16
6.10 Fe(II) D1 1.33 0.28 3.37 21 QSD1 1.30 3.19 0.33 64
D2 1.30 0.44 2.91 56 1.30 2.61 0.27 12
Fe(III) D3 0.43 0.57 0.70 23 QSD2 0.42 0.73 0.46 24
7.08 Fe(II) D1 1.34 0.33 3.27 18 QSD1 1.31 3.13 0.29 31
D2 1.29 0.44 2.87 39 1.31 2.67 0.70 25
Fe(III) D3 0.48 0.49 0.63 43 QSD2 0.47 0.70 0.40 44
8.66 Fe(II) D1 1.27 0.27 3.10 45 QSD1 1.27 3.10 0.18 44
D2 1.27 0.38 2.75 48 1.27 2.79 0.35 48
Fe(III) D3 0.39 0.36 0.55 7 QSD2 0.43 0.70 0.40 8
4.91 Fe(II) D1 1.29 0.38 2.96 49 QSD1 1.29 3.00 0.71 17
D2 1.28 0.32 2.52 34 1.29 2.76 0.33 64
Fe(III) D3 0.42 0.40 0.68 17 QSD2 0.45 0.80 0.45 19
The temperature of the analyses was 77 K. The pH of the suspension is given for each spectrum. Two methods of model fitting were used: the MOSFIT program (unpublished program, Teillet and Varret, Universite´ du Maine, Le Mans, France) and the QSDs method ofRancourt and Ping (1991).d(mm s1) isomer shift with respect to metallica-Fe(0) at room temperature;DE
Q(mm s1) quadrupole splitting, RA (%) relative abundance;C(mm s1) full
width at half maximum.ÆCSæ(mm s1) average centre shift,ÆQSæ(mm s1) the average quadrupolar splitting,r
sBðOHÞðO2Þ2Fe2þ()sBðeÞ ðOHÞðO2Þ2Fe3þ ð2Þ
In reaction (2), one electron is transferred from the sorbed Fe(II) to a nearby surface or near-surface site on or in the montmorillonite. The complex that receives the electron is denoted (e)* and will be some relatively receptive center, as determined by local bond valence and local electronega-tivity conditions. Reaction (2) aloneis probably not ener-getically favored (given the ionization energy of Fe(II)), but we imagine it to be accompanied by a local structural and/or chemical adjustment that greatly stabilizes sorbed Fe(III), a small ion, relative to sorbed Fe(II), a large ion, such that the net change in free energy per sorption/oxida-tion event is negative for the given sorpsorption/oxida-tion-site/electron- sorption-site/electron-acceptor-site complex. The required structural-chemical adjustment can be represented as
sBðeÞ ðOHÞðO2Þ2Fe3þ ()s
BðeÞ ðO2Þ3Fe3þþHþ
aq ð3Þ
Here, we bias our choice among possible structural-chemi-cal adjustments to involve a surface proton loss. This is be-cause we believe that sorbed Fe(III) will be favored, compared to sorbed Fe(II) with the first coordination shell proton removed for two reasons: (1) at the s„OH group, the H+–Fe(III) repulsion will be greater than the H+–Fe(II) repulsion and (2) the spherically symmetric and predomi-nantly ionic bonding Fe(III) will be able to more easily ad-just a three surface-bound coordinating O2 chelate than
the largely covalent bonding ferrous cation that will more easily adjust solution-side first shell anions than surface-bound ones to its covalent (bond angle and distance) requirements. This choice is also consistent with our obser-vation that the fraction of sorbed Fe present as Fe(III) increases with increasing pH in the sorption plateau region (Figs. 3 and 7). Of course, conversely, it is also clear that some surface complexation sites will have first shell coordi-nating anions that sterically favor Fe(II) and that these sites will form relatively stable Fe(II) complexes, which would be statistically the last ones to oxidize upon intro-duction of dissolved oxygen in the system. All our discus-sion refers to a sorbant that displays a large array of complexation sites, as previously described to be the case for montmorillonite (Tournassat et al., 2004).
Since the synthetic montmorillonite is assumed to be a perfect insulator, the question of the nature of the oxidant site remains and one can not exclude the possibility of re-dox reaction with the only one electron acceptor in the sys-tem i.e. water. The stabilization of sorbed Fe(III) is then perhaps linked to the reduction of water via the following reaction, then replacing Eq.(3):
sBðeÞ ðOHÞðO2Þ2Fe3þþ2H
2O
()sBðO2Þ3Fe3þðOHÞ
...
2ðH2Þ0:5þ2Hþaq ð4Þ
Reactions (2) and (3) or (2) and (4) together represent our proposed reversible anoxic-environment sorbed-Fe(II) oxi-dation mechanism. Such oxioxi-dation will advance if there is a
Fig. 6. (A) Evolution of the Fe(II) component from RECOIL fits of the full Mo¨ssbauer signal: Fe(II) Mo¨ssbauer spectrum (left side) and of the quadrupolar splitting distribution given as distribution probabilities (p(D) in mm s1) as a function of quadrupolar splitting (D in mm s1). (B)
Evolution of the overall average quadrupolar splittingÆŒDŒæof the Fe(II) site (filled triangles) and of the overall standard deviation width of the entire Fe(II) quadrupolar splitting distribution QSD (filled circles) as a function of pH.
net free energy gain per Fe oxidation event. Since one or two protons are produced, part of the free energy change involves the proton activity (pH). Increasing pH drives the overall reaction to the right, as observed. The other component of the change in free energy for the overall oxi-dation reaction (reactions 1 and 2 together) is equal to the difference in binding (and ionic) free energy between the s„(OH)(O2)2Fe(II) and s„(e)*(O2)3Fe(III) complex-es, including the Fe ionization energy. This difference,DFs, depends on bonding strengths and has electrostatic, vibra-tional, entropic, and steric contributions. The steric contri-bution is largely the so-called ligand field stabilization energy. DFs, therefore, is strongly dependent on the local structure of the specific bonding site, including its position in nanovoids, local electronegativities, exact coordinating anion positions and stiffnesses, etc. We propose that the montmorillonite edge sites include a population of sites that have DFs< 0 and that these sites, therefore, cause the net observed Fe(II) to Fe(III) oxidations.
The numerical applicability of such a model was tested using a very simple complexation model. As already stated, the discussion was in the context of a sorbant that displays a large array of complexation sites. A model with two ‘‘strong’’ sites (s1, s2) was then chosen where surface pro-cesses are possible following Eqs.(2) and (4). A one strong site model was also tested but was proved to be inefficient in reproducing the experimental data. In addition, cation exchange and sorption on a weak site (w) were also consid-ered. Fe–Ca and Fe–Na cation exchange selectivity con-stants in 0.1 mol/L chloride anionic background were taken from the literature (Charlet and Tournassat, 2005,
Table 2).
Given our ignorance of the sorption sites localization and the common use of a non-electrostatic model for clay minerals (Bradbury and Baeyens, 1997, 1999, 2005b; Tour-nassat et al., 2004), we express Fe(II) sorption equations in
terms of reaction with a neutral s„3+(OH)3site. Eq.(1)is
These reactions for sites s1and s2occur at a pH value lower than where the main edge of the curve is observed (Fig. 3). They are therefore very poorly constrained concerning the Fe2+/H+balance and the sorption constant. However, the choice of Fe2+/H+ balance for Fe sorption reaction on ‘‘weak’’ sites is crucial in order to describe as well as possi-ble the slope of the sorption edge at pH 7–8. It was found that a balance of 3H+ for one Fe2+was the best solution corresponding, for example, to a hydroxylated bidentate complex („w2O2FeOH) or a bihydroxylated mon-odentate complex ðBwOFeðOHÞ2Þ. The arbitrarily select-ed model corresponds to a hydroxylatselect-ed bidentate complex:
2BwOHþFe2þþH2O
()Bw2O2FeOHþ3Hþ logKwsorp ð6Þ
Eqs.(2) and (4)were then combined into a single equation identical for the three strong sites:
sB3þðOHÞðO2Þ2Fe2þþ2H2O
Here, we have arbitrarily chosen to keep H2on the Fe sorp-tion site and to consider that it is not further released in solution given the absence of H2 concentration mea-surements.
The total amount of strong sites can be deduced from experimental results by considering that weak sites did not oxidize sorbed Fe(II) and that strong sites completely oxidized Fe(II) into Fe(III) at pH 8.66. Considering Fe dilution steps during the experiment (by alkali solution additions), one calculates that the sorption of Fe(II) at pH 8.66 corresponds to a total amount of 64 mmol Fe/ kgclay. Seven percentage of this amount was oxidized and then the total amount of strong sites was4.5 mmol/kgclay. The amount of strong sites can be also be deduced from the difference between the experimental curve and the modeled amount of Fe(II) that underwent cation exchange given that no parameter is needed to be adjusted for cation ex-change reaction modeling (Fig. 8). This difference led to a value of4.2 ± 1 mmol/kgclay, in agreement with previous value. The logKssorp1;s2 constant were adjusted so that Fe
2+
start to be adsorbed below pH 2 in order to explain the flat shape of the sorption edge curve between pH 2 and pH 6. The relative amounts of sites s1 and s2 were then fitted thanks to the evolution of the percentage of Fe(III) on the montmorillonite surfaces as a function of pH (Fig. 7). One cannot fail to observe that the strong site density in this model is very similar to the density of the so called strong sites in the Bradbury and Baeyens studies
Table 2
Cation exchange parameters for Ca–Fe and Na–Fe exchange in 0.1 mol l1
chloride anionic background
Exchange reaction logKGTa
BXNaþHþ()
The CEC value was considered to be 0.63 eq/kg at pH 7 according to the Cs–Li method results. The permanent structural charge (Cs result) was found to be 0.44 mol/kg.
a
Note that the selectivity coefficients are given in the Gaines and Thomas convention (Sposito, 1981, log KGT) and not in the Vanselow
convention (log Kv) as claimed previously in Charlet and Tournassat,
(2 mmol/kg, Bradbury and Baeyens, 1997, 1999, 2002, 2005a,b). The complete set of modeling parameters is given inTables 2–4.
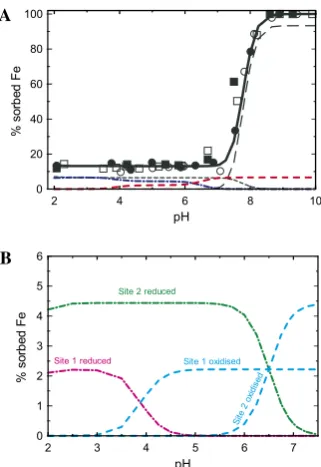
Figs. 7 and 8show that both solution analysis and spec-trometric results are well reproduced by the model. In the pre-edge pH region (from pH 2 to pH 7.5), the Fe(II)
uptake is equivalent between Fe(II) exchanged under the form of Fe2+ (15% of the signal) and FeCl+ (35% of the signal) and strongly sorbed in Fe(II) or Fe(III) form (50% of the signal). Above pH 7.5, the exchanged frac-tion of sorbed Fe(II) decreases drastically and the uptake is dominated by ‘‘weak’’ sorption sites.
Once the main contributors of Fe(II) uptake are known, it is possible to create a Mo¨ssbauer spectra decomposition model, taking into account all of this information (Table 5).
First, the spectrum at pH 4.03 was decomposed into five doublets corresponding to exchanged Fe2+, exchanged FeCl+, Fe(II) sorbed on strong site 1, Fe(II) sorbed on strong site 2 and Fe oxidized on strong sites. Mo¨ssbauer parameters for Fe2+and FeCl+in cation exchanged posi-tion were taken from Charlet and Tournassat, 2005.
Fig. 9(bottom) shows that this model allows reproducing the Mo¨ssbauer spectrum with the proportion between dif-ferent Fe sorption sites comparing well with those given by the chemical sorption model (Table 5, comparison of the two last rows). Spectra at higher pH were then fitted by adding fitting component when necessary. Again, the re-sult from this fit compares well with the rere-sult from the chemical model (Fig. 9 and Table 5) giving sense to our modeling approach. At pH 8.66, the need to use at least 3 Mo¨ssbauer components is likely to represent the com-plexity in structure and chemistry of the clay edge sites. One can also note that the component weak 3 (Table 5, pH 8.66) is very similar to the one used for exchanged Fe2+ and for Fe(OH)2(s) (Fig. 2): it is then probable that part of specifically but weakly sorbed Fe(II) forms outer-sphere complexes, surface or bulk precipitate at pH 8.66. The two latter interpretations are preferred given the exper-imental conditions and the Fe(OH)2(s) solubility curve (Fig. 3).
The nature of the strongly sorbing sites is still unknown. However, given their fitted amount (4.2 mmol/kg), it is possible to draw some hypotheses based on the knowledge of the structure and chemistry of the montmorillonite edg-es. Analyzing the edge structure proposed by Tournassat et al. (2004), one notes that tridendate sorption sites as pro-posed in this paper can be constituted by the combination of the following elementary sites:
•One MeOh–O site associated to one MeOh–O–MeT site (with the same MeOH cation, MeOH= Al or Mg) and either one MeOh–O–MeOH site or one MeOh2–O–MeT site;
A
B
Fig. 8. (A) Comparison of experimental sorption data with the proposed model. Full line: total sorbed Fe(II). Black short dash line: cation exchanged Fe(II). Black long dash line: Fe(II) sorbed on weak sites. Blue dash-dot line: Fe(II) sorbed on strong sites. Red dash line: Fe(III) sorbed on strong sites. (B) Details in the pH < 7.5 zone: decomposition of strong sites s1 and s2 contributions. Blue dash-dot
line: Fe(III) sorbed on strong sites s1and s2. Pink and green dash lines:
Fe(II) sorbed on strong sites s1 and s2. Colors on Fig. 8B refer to the
colors used in Fig. 9.
Table 3
Site amounts for edge sorption sites and exchange sites (in mmol/kgclay)
Name of site Site amount (mmol/kgclay)
s1
„3+(OH)3 1.4
s2
„3+(OH)3 2.8
w„OH 80
X(exchange sites) 630 (pH 7)–440 (pH 2)
The CEC value was measured to be 0.63 eq/kg at pH 7 according to the Cs–Li method results (Anderson and Sposito, 1991). The permanent structural charge was found to be 0.44 mol/kg. This last value was applied to the model, since specific sorption and oxidation start at low pH.
Table 4
Fe affinity constants for strong and weak sites and Fe(II) oxidation constants on strong sites
Reaction logK
Fe2þ
þs1B3þðOH
Þ3()s1B3þðOHÞðO2Þ2Fe2þþ2Hþaq 1
Fe2þ
þs2B3þðOH
Þ3()s2B3þðOHÞðO2Þ2Fe2þþ2Hþaq 1
s1B3þðOH ÞðO2
Þ2Fe2þþ2H2O()s1B3þðO2Þ3Fe3þðOHÞ...2ðH2Þ0:5þ2Hþaq 7.8
s2B3þðOH ÞðO2
Þ2Fe2þþ2H2O()s2B3þðO2Þ3Fe3þðOHÞ...2ðH2Þ0:5þ2Hþaq 13
2BwOHþFe2þþH
• Two associated MeOh–O sites associated to either one MeOh–O–MeOHsite or one MeOh2–O–MeTsite;
• Two MeOh–O–MeT sites associated to one MeOh2–O– MeTsite.
By using equal probability combination rules as given in
Tournassat et al. (2004)with the structural formula of our synthetic montmorillonite and with a edge specific surface area of 8 m2/g (Tournassat et al., 2003), one can calculate that Mg cations in octahedral position and fulfilling the
above requirement in tridentate sites are present in the fol-lowing amounts:2 mmol/kg for one MgOh–O site associ-ated to one MgOh–O–Si site and one MeOh–O–MeOH site or one MeOh2–O–MeT site (Me = Al or Mg),0.9 mmol/ kg for two associated MgOh–O sites associated to either one MgOh–O–MeOH site or one MeOh2–O–MeT site and 2 mmol/kg for two MgOh–O–Si sites associated to one MeOh2–O–MeTsite. Assuming the first reactive site config-uration given above, one obtains the following reaction scheme:
Mg
Al Si
OH+0.33
OH2+0.83
OH2+0.33 + Fe+2 Mg Fe+2 + 2 H+ (8)
Al Si
O-0.67
OH2+0.83 OH-0.67
Table 5
Mo¨ssbauer hyperfine parameters adjusted with constraints from the chemical modeling approach
pH Site (color inFig. 9) d C DEQ RA (%) Comparison with chemical modeling (%)
4.03 Dexch FeCl+(red) 1.38 0.30 3.43 36 36
Dexch Fe2+(blue) 1.40 0.28 3.10 16 14
Dstrong_red1(pink) 1.39 0.32 3.00 15 6
Dstrong_red2(green) 1.23 0.32 2.97 22 34
Dstrong_ox(cyan) 0.43 0.41 0.65 11 11
5.01 Dexch FeCl+(red) 1.36 0.34 3.41 36 35
Dexch Fe2+(blue) 1.34 0.28 3.02 16 14
Dstrong_red2(green) 1.30 0.26 2.74 30 34
Dstrong_ox(cyan) 0.49 0.59 0.65 17 17
6.10 Dexch FeCl+(red) 1.34 0.34 3.33 36 35
Dexch Fe2+(blue) 1.33 0.28 3.01 16 14
Dstrong_red2(green) 1.32 0.32 2.72 23 28
Dstrong_ox(cyan) 0.47 0.55 0.65 25 23
7.08 Dexch FeCl+(red) 1.38 0.34 3.18 27 32
Dexch Fe2+(blue) 1.24 0.28 3.04 12 13
Dstrong_ox(cyan) 0.49 0.51 0.65 47 45
Dstrong_red2 — — — 0 2
Dweak1(black) 1.32 0.30 2.67 14 8
8.66 Dexch FeCl+ — — — 0 0
Dexch Fe2+ — — — 0 0
Dstrong_ox(cyan) 0.46 0.53 0.65 9 7
Dweak1(black) 1.39 0.30 2.77 21
Dweak2(black) 1.18 0.30 2.95 30 93
Dweak3(black) 1.33 0.28 3.04 40
The temperature of the analyses is 77 K. The pH of the suspension is given for each spectrum.d(mm s1) isomer shift with respect to metallica-Fe(0) at
room temperature;DEQ(mm s1) quadrupole splitting, RA (%) relative abundance;C(mm s1) full width at half maximum. The subscripts exch FeCl+,
exch Fe2+, strong_red, strong_ox and weak refer to the modeling sorption site given inTable 4.
+ H2O Mg + H+ (9)
Al Si
O-0.67
OH2+0.83 OH-0.67 Mg
Al Si
O-0.67
OH2+0.83
OH-0.67 Fe+2 Fe+2(OH)
Reaction (8) produces a near-neutral complex (the for-mal charge is here +0.5, but a more accurate account of bond valences might yield even a neutral surface complex). Once the pH increases, the Fe(II) atom of the complex is hydrolyzed producing a charge imbalance of the surface complex (reaction (9)). This imbalance is further compen-sated by the oxidation of sorbed Fe(II) into sorbed Fe(III) (reaction (10)). The oxidation reaction can then be seen as one way the system responds to maintain a stable
sur-face species in the sur-face of an instability provoked by hydrolysis.
At the moment it is not possible to say if the good agree-ment between the fitted amount of strong site in this study and the calculated amount of most electronegative sites after the structural model by Tournassat et al. (2004) is as a coincidence or not. In a future study, molecular dynamics combined with electronic structure calculations will help to confirm (or infirm) the mechanism that we
Fig. 9. Mo¨ssbauer spectra atT= 77 K shown together with hyperfine contributions obtained using the MOSFIT approach combined to constrains from chemical sorption model. Hyperfine parameters are given inTable 5together with the color/site relation. Wide black continuous line is the sum of all of the contributions to the spectra.
+ 0.5 H2
(10) Mg
Al Si
O-0.67
OH2+0.83
OH-0.67 Fe+3(OH)
2-2 Mg
Al Si
O-0.67
OH2+0.83 OH-0.67 Fe+2(OH)
-+ H2O
propose on these types of sites and to refine the H+/Fe sto-chiometry of the reaction.
4. Conclusions
Based on wet analytical chemistry and Mo¨ssbauer spectrometry, we have shown that Fe(II) has a peculiar behavior when sorbed to the surfaces of an iron-free syn-thetic montmorillonite. Fe(II) undergoes classical uptake processes by cation exchange (in the form of Fe2+ but also FeCl+) and specific sorption. The main sorption edge occurs at pH7 whereas the low pH plateau region is quantitatively dominated by cation exchange processes. In addition, Fe(II) is sorbed at very low pH on specific sites and undergoes partial to total oxidation as a func-tion of pH in absence of any oxidant in the solufunc-tion other than H2O. This oxidation is fully reversible point-ing out (with other experimental evidences) that this oxidation is not due to an accidental oxidation by O2 impurities. The nature of the sites responsible for this redox phenomenon is still unclear. The only reliable elec-tron acceptor seems to be water itself with production of H2that needs to remain in the vicinity of the clay surfac-es in order to ensure the reversibility of the redox reaction.
A simple non-electrostatic sorption model reproduces accurately both wet chemical as well as Mo¨ssbauer spectro-metric results. The model considers, in agreement with hyperfine parameters, the Fe(II) to be present on the three types of clay edge sites. A weak site where, like in the interlayer, oxidation of Fe(II) does not occur. Two strong sites present at a concentration of 1.4 and 2:8 mmol kgclay1 , where oxidation occurs around pH 4.0 and 6.5, respective-ly. The overall importance of oxidized iron in the 57Fe Mo¨ssbauer signal decreases at higher pH values, as Fe(II) is being adsorbed on weak edge sites around pH 7.5. These observations further support the concept introduced by theoretical calculations of redox interactions with miner-al–mineral non equivalent sites (Becker et al., 2001). This study points out that most of the reactivity of a surface is related to minority sites, such as edge or defect sites. It is estimated that as little as 0.6% of the total surface sites is involved in Fe redox reaction in the present study.
Both the exact nature of the edge site where the redox reactions occur, the nature of the electron storage capacity, here assumed to be sorbed hydrogen gas molecules, and the exact mechanism of electron transfer deserve further inves-tigations. In a companion paper (Charlet et al., Submitted), the reducing capacity of Fe(II) sorbed on iron-free synthet-ic montmorillonite edge sites towards Se(IV) is demonstrat-ed, and found to be in agreement with the model developed in the present paper.
Acknowledgments
This research is part of a Ph.D. study and a post-doc-torial study initiated, monitored and supported by the
ANDRA (French National Radioactive Waste Manage-ment Agency), (1) in the framework of its program on the geochemical behavior of bentonite engineered barriers under the supervision of Dr. N. Michau and (2) in the framework of the 6th PCRD Euratom FUNMIG pro-gram under the supervision of Dr. S. Altmann. Support to DGR was provided by the Natural Sciences and Engi-neering Research Council of Canada. Three anonymous reviewers and A.E. Pr. Sverjensky are acknowledged for their comments on the manuscript. Finally, we thank Pr. Garrison Sposito for helpful discussion on the oxida-tion mechanism.
Associate editor:Dimitri A. Sverjensky
References
Akcay, H., 1998. Aqueous speciation and pH effect on the sorption behavior of uranium by montmorillonite. J. Radioanal. Nucl. Ch.237, 133–137.
Anderson, S.J., Sposito, G., 1991. Cesium-adsorption method for mea-suring accessible structural surface charge. Soil Sci. Soc. Am. J.55, 1569–1576.
Baeyens, B., Bradbury, M.H., 1997. A mechanistic description of Ni and Zn sorption on Na-montmorillonite. Part I: Titration and sorption measurements. J. Contam. Hydrol.27, 199–222.
Balzer, W., 1982. On the distribution of iron and manganese at the sediment/water interface: thermodynamic versus kinetic control. Geo-chim. CosmoGeo-chim. Acta.46, 1153–1161.
Becker, U., Rosso, K.M., Hochella, J., Michael, F., 2001. The proximity effect on semiconducting mineral surfaces: a new aspect of mineral surface reactivity and surface complexation theory?Geochimi. Cosmo-chim. Acta.65, 2641–2649.
Berner, R.A., 1971.Principles of Chemical Sedimentology. McGraw-Hill Companies, pp. 240.
Bott, M. 2002. Iron sulfides in Baldeggersee during the last 8000 years: formation processes, chemical speciation and mineralogical constrains from EXAFS spectroscopy, Thesis ETH Zu¨rich.
Bradbury, M.H., Baeyens, B., 1997. A mechanistic description of Ni and Zn sorption on Na-montmorillonite. Part II: modeling. J. Contam. Hydrol.27, 223–248.
Bradbury, M.H., Baeyens, B., 1999. Modeling the sorption of Zn and Ni on Ca-montmorillonite. Geochimi. Cosmochim. Acta.63, 325–336. Bradbury, M.H., Baeyens, B., 2002. Sorption of Eu on Na- and
Ca-montmorillonites: experimental investigations and modeling with cation exchange and surface complexation. Geochim. Cosmochim. Acta.66, 2325–2334.
Bradbury, M.H., Baeyens, B., 2005a. Experimental measurements and modeling of sorption competition on montmorillonite. Geochim. Cosmochim. Acta.69, 4187–4197.
Bradbury, M.H., Baeyens, B., 2005b. Modelling the sorption of Mn(II), Co(II), Ni(II), Zn(II), Cd(II), Eu(III), Am(III), Sn(IV), Th(IV), Np(V) and U(VI) on montmorillonite: Linear free energy relationships and estimates of surface binding constants for some selected heavy metals and actinides. Geochim. Cosmochim. Acta.69, 875–892.
Charlet, L., Bosbach, D., Peretyashko, T., 2002. Natural attenuation of TCE, As, Hg linked to the heterogeneous oxidation of Fe(II): an AFM study. Chem. Geol.190, 303–319.
Charlet, L., Liger, E., Gerasimo, P., 1998a. Decontamination of TCE- and U-rich waters by granular iron: role of sorbed Fe(II). J. Environ. Eng.
124, 25–30.
Charlet, L., Schindler, P.W., Spadini, L., Furrer, G., Zysset, M., 1993. Cation adsorption on oxides and clays: The aluminum case. Aquat. Sci.55, 1015–1621.
Charlet, L., Silvester, E., Liger, E., 1998b. N-compound reduction and actinide immobilisation in superficial fluids by Fe(II): the sur-face = FeIIIOFeIOH0 species, as major reductant. Chem. Geo. 151,
85–93.
Charlet, L., Tournassat, C., 2005. Fe(II)-Na(I)-Ca(II) cation exchange on montmorillonite in chloride medium; evidence for preferential clay adsorption of chloride – metal ion pairs in seawater. Aquat. Geochem.
11, 115–137.
Criaud, A., Fouillac, C., 1986a. Etude des eaux thermomine´rales carbogazeuses du Massif Central franc¸ais. I. Potentiel d’oxydo-re´duction et comportement du fer. Geochim. Cosmochim. Acta. 50, 525–533.
Criaud, A., Fouillac, C., 1986b. Etude des eaux thermomine´rales carbogazeuses du Massif Central franc¸ais. II. Comportement de quelques me´taux en trace, de l’arsenic, de l’antimoine, et du germa-nium. Geochim. Cosmochim. Acta.50, 1573–1582.
Davison, W., Phillips, N., Tabner, B.J., 1999. Soluble iron sulfide species in natural waters: reappraisal of their stoichiometry and stability constants. Aquat. Sci.61, 23–43.
Drever, J.I., 1971. Magnesium-iron replacement in clay minerals in anoxic marine sediments. Science172, 1334–1336.
Emerson, S., 1976. Early diagenesis in anaerobic lake sediments: chemical equilibria in intersticial waters. Geochim. Cosmochim. Acta.
40, 925–934.
Emerson, S., Jahnke, R., Bender, M., Froelich, P., Klinkhammer, G., Bowser, C., Setlock, G., 1980. Early diagenesis in sediments from the eastern equatorial pacific. I. pore water nutrient and carbonate results. Earth Planet. Sci. Lett.49, 57–80.
Evans, R.J., Rancourt, D.G., Grodzicki, M., 2005. Hyperfine electric field gradient tensors at Fe2+ sites in octahedral layers: Toward
under-standing oriented single-crystal Mo¨ssbauer spectroscopy measure-ments of micas. Am. Mineral.90, 1540–1555.
Fletcher, P., Sposito, G., 1989. The chemical modeling of clay/electrolyte interactions for montmorillonite. Clay Miner.24, 375–391.
Hofstetter, T.B., Heijman, C.G., Haderlein, S.B., Holliger, C., Schwar-zenbach, R.P., 1999. Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface con-ditions. Env. Sci. Technol.33, 1479–1487.
Hofstetter, T.B., Neumann, A., Schwarzenbach, R.P., 2006. Reduction of Nitroaromatic Compounds by Fe(II) Species Associated with Iron-Rich Smectites. Env. Sci. Technol.40, 235–242.
Hofstetter, T.B., Schwarzenbach, R.P., Haderlein, S.B., 2002. Reactivity of Fe(II) species associated with clay minerals. Env. Sci. Technol.37, 519–528.
Kamei, G., Oda, C., Mitsui, S., Shibata, M., Shinozaki, T., 1999. Fe(II)-Na ion exchange at interlayers of smectite: adsorption-desorption experiments and a natural analogue. Eng. Geol.54, 15–20.
Klausen, J., Tro¨ber, S.P., Haderlein, S.B., Schwarzenbach, R.P., 1995. Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspension. Env. Sci. Technol.29, 2396–2404.
Liger, E., Charlet, L., Van Cappellen, P., 1999. Surface catalysis of uranium (VI) reduction by iron(II). Geochim. Cosmochim. Acta.63, 2939–2955.
Naftz, D.L., Feltcorn, E.M., Fuller, C.C., Wilhelm, R.G., Davis, J.A., Morrison, S.J., Freethey, G.W., Piana, M.J., Rowland, R.C., and Blue, J.E. 2000. Field demonstration of permeable reactive barriers to remove dissolved uranium from groundwater, Fry Canyon, Utah, Report No. EPA 402-C-00-001.
Parkhurst, D.L., Appelo, C.A.J. 1999. User’s guide to phreeqc - a computer program for speciation, batch-reaction, one-dimensional
transport, and inverse geochemical calculations, USGS Report No. 99–4259.
Pecher, K., Haderlein, S.B., Schwarzenbach, R.P., 2002. Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides. Env. Sci. Technol.36, 1734–1741. Peretyazhko, T., Sposito, G., 2005. Iron(III) reduction and phosphorous
solubilization in humid tropical forest soils. Geochim. Cosmochim. Acta.69, 3643–3652.
Postma, D., 1982. Pyrite and siderite formation in brackish and freshwater swamp sediments. Am. J. Sci.282, 1151–1183.
Rancourt, D.G., 1994a. Mo¨ssbauer spectroscopy of minerals I. Inade-quacy of Lorentzian-line doublets in fitting spectra arising from quadrupole splitting distributions. Phys. Chem. Miner.21, 244–249. Rancourt, D.G., 1994b. Mo¨ssbauer spectroscopy of minerals II. Problem
of resolving cis and trans octahedral Fe2+sites. Phys. Chem. Miner.21,
250–257.
Rancourt, D.G., 1998. Mo¨ssbauer spectroscopy in clay science. Hyperfine Interact.117, 3–38.
Rancourt, D.G., Ping, J.Y., 1991. Voigt-based methods for arbitrary-shape static hyperfine parameter distributions in Mo¨ssbauer spectros-copy. Nucl. Instrum. Methods Phys. Res. Sect. B (NIMB)58, 85–97. Rancourt, D.G., Ping, J.Y., Berman, R.G., 1994. Mo¨ssbauer spectroscopy
of minerals III. Octahedral-site Fe2+quadrupole splitting distributions
in the phlogopite-annite series. Phys. Chem. Miner.21, 258–267. Rancourt, D.G., Thibault, P.J., Mavrocordatos, D., Lamarche, G., 2005.
Hydrous ferric oxide precipitation in the presence of nonmetabolizing bacteria: Constraints on the mechanism of a biotic effect. Geochim. Cosmochim. Acta.69, 553–577.
Reinholdt, M., Mie´he´-Brendle´, J., Delmotte, L., Tuillier, M.-H., de Dred, R., Corte`s, R., Flank, A.-M., 2001. Fluorine route synthesis of montmorillonites containing Mg or Zn and characterization by XRD, thermal analysis, MAS NMR, and EXAFS spectroscopy. Eur. J. Org. chem.2001, 2831–2841.
Sposito, G., 1981.The thermodynamics of soil solution. Oxford University Press.
Strathmann, T.J., Stone, A.T., 2003. Mineral surface catalysis of reactions between FeIIand oxime carbamate pesticide. Geochim. Cosmochim. Acta.67, 2775–2791.
Thompson, A., Chadwick, O.A., Rancourt, D.G., Chorover, J., 2006. Iron-oxide crystallinity increases during soil redox oscillations. Geo-chim. CosmoGeo-chim. Acta.70, 1710–1727.
Tournassat, C., Ferrage, E., Poinsignon, C., Charlet, L., 2004. The titration of clay minerals. Part II. Structural-based model and implications for clay reactivity. J. Colloid Interf. Sci.273, 234–246. Tournassat, C., Neaman, A., Villie´ras, F., Bosbach, D., Charlet, L., 2003.
Nanomorphology of montmorillonite particles: Estimation of the clay edge sorption site density by low-pressure gas adsorption and AFM observations. Am. Mineral.88, 1989–1995.
Turner, D.R., Pabalan, R.T., Bertetti, F.P., 1998. Neptunium(V) sorption on montmorillonite: an experimental and surface complexation mod-eling study. Clay. Clay Miner.46, 256–269.
Viollier, E., Inglett, P.W., Hunter, K., Roychoudhury, A.N., Van Cappellen, P., 2000. The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters. Appl. Geochem.15, 785–790. Wehrli, B., 1990. Redox reactions of metal ions at mineral surfaces. In:
Stumm, W. (Ed.),Aquatic chemical kinetics: reaction rates of processes in natural waters. Wiley, pp. 311–366.
Zachara, J.M., Smith, S.C., 1994. Edge complexation reactions of cadmium on specimen and soil-derived smectite. Soil Sci. Soc. Am. J.58, 762–769.
![Fig. 1. X-ray diffraction patterns of the Ca0.3synthetic montmorillonite (grey diffractogram) and the Ca[(Al1.4Mg0.6)Si4O10 (OH,F)2]0.14 (Al1.61Mg0.24 FeIII0:15 FeII0:02) (Si3.98Al0.02) O10(OH)2 MX80 montmorillonite (blackdiffractogram).](https://thumb-ap.123doks.com/thumbv2/123dok/3763533.1820534/2.595.319.532.570.701/diraction-patterns-synthetic-montmorillonite-diractogram-feiii-montmorillonite-blackdiractogram.webp)