APRIL 2016 Volume 28 Number 4
FORMULATION
Container Selection for Biologic FormulationsPEER-REVIEWED
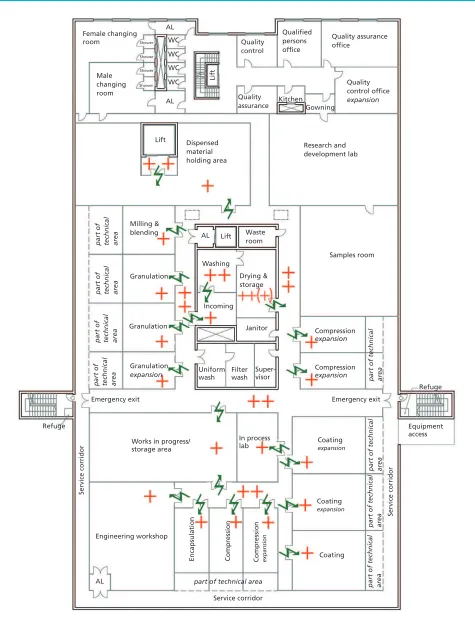
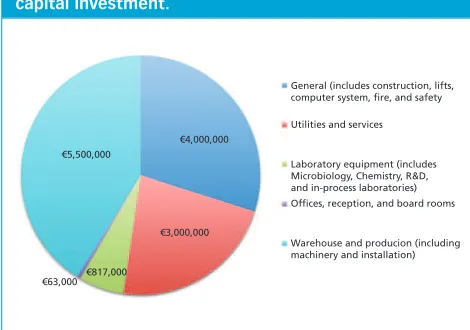
Establishing an Eco-Friendly Facility in Malta
QUALITY
FMEA for
Filter Integrity TestingAdvances for
Bio/Pharma
That’s why we’ve added Celsis
®. to our family of industry-leading brands
. Customers have long trusted our
Endosafe
®endotoxin testing and
Accugenix
®microbial identification solutions to bring their products to market safely and efficiently.
Celsis
®now rounds out the trio, offering rapid microbial detection systems that make us unmatched in our ability to support
your micro QC programs, every step of the way, learn more at
www.criver.com/celsis.
2 CHEERS
2 COINS IN A FOUNTAIN
2 FRENCH HENS
2 LITTLE PIGS
GOLDILOCKS AND THE 2 BEARS
2 PIECE SUIT
2 STOOGES
2 BLIND MICE
2 DIMENSIONAL
2 MUSKETEERS
2 TENORS
Cover: Resolution Productions/ Getty Images
Art direction: Dan Ward
April 2016
Advances for Bio/Pharma Analytical Laboratories Today’s analytical laboratory equipment reflects the realities of downsizing, outsourcing,and the need for speed.
Container Selection for Biologic Formulations Choosing the right container and container closure system is crucial for ensuring product quality, safety, and efficacy of a biologic formulation.
Headspace Moisture Analysis for Determination of Residual Moisture Content in Lyophilized Pharma-ceutical Products
Headspace moisture analysis is a rapid non-destructive analytical method that may potentially address the limitations of traditional methods used for residual moisture determination.
Failure Mode Effects Analysis for Filter Integrity Testing Understanding the risks associated with FMEA is crucial in lot release testing.
Editor’s Comment
The Repercussions of Data Integrity Violations
Outsourcing Review CMC Development is Hot
European Regulatory Watch
Europe Moves Forward on Anticounterfeiting Measures
US Regulatory Watch
Vaccine Development Faces Urgency and Challenges
API Synthesis & Manufacturing
Conjugation Chemistry with Highly Potent Compounds
Packaging Forum Blister Pack Optimization
Troubleshooting
Understanding Measurement Uncertainty in Weighing
Ad Index
Concept Design for Establishing an Eco-Friendly Pharmaceutical Production Facility in Malta The authors discuss the concept design of a versatile, sustainable, small-scale facility in Malta.
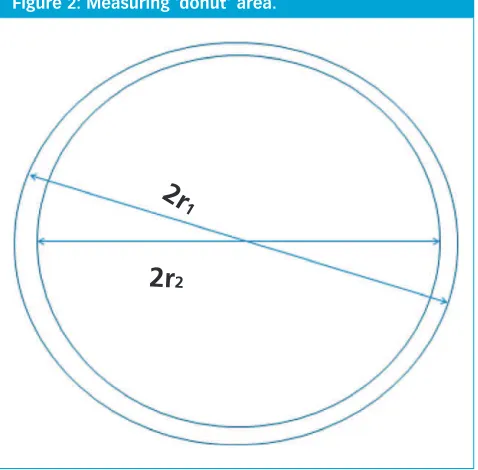
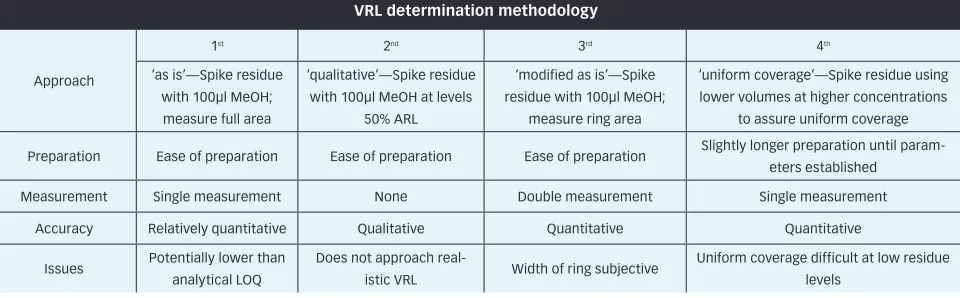
Ruggedness of Visible Residue Limits for Cleaning Validation, Part IV: Defining Visible Residue Limits to be Practical, Achievable, and Verifiable Visible residue limits have been shown to be a valuable tool in a validated cleaning validation programme.
Join the Pharmaceutical Technology Europe group on LinkedIn™* and start discussing the issues that matter to you with your peers.
Go to
* The linkedIn logo is a registered trademark of LinkedIn Corporation and its afi liates in the United States and/or other countries
Pharmaceutical Technology Europe is the authoritative source of peer-reviewed research and expert analyses for scientists, engineers, and managers engaged in process development, manufacturing, formulation and drug delivery, API synthesis, analytical technology and testing, packaging, IT, outsourcing, and regulatory compliance in the pharmaceutical and biotechnology industries.
Advancing Development & Manufacturing
PharmTech.com
PharmTech Europe Cynthia A. Challener, PhD Global Correspondent
Cheshire CH65 9HQ, United Kingdom Tel. +44 151 353 3500
Fax +44 151 353 3601
UBM Americas:
EVP & Senior Managing Director, Life Sciences Group
Tom Ehardt Senior VP, Finance Tom Mahon
EVP & Managing Director, UBM Medica
Georgiann DeCenzo EVP, Strategy & Business Development
Mike Alic
VP & Managing Director, Pharm/ Science Group
Dave Esola
VP & Managing Director, CBI/IVT Johanna Morse
VP & Managing Director, Veterinary Group Becky Turner Chapman VP, Marketing & Audience Development
Vice-President, R&D Instrumentation & Control Sartorius AG
Rafael Beerbohm
Director of Quality Systems Boehringer Ingelheim GmbH
Phil Borman
Manager, GlaxoSmithKline
Rory Budihandojo
Director, Quality and EHS Audit Boehringer-Ingelheim
Professor in Pharmaceutics and Pharmaceutical Technology Trinity College Dublin, Ireland
Deirdre Hurley
Senior Director, Plant Helsinn Birex Pharmaceuticals Ltd.
Makarand Jawadekar
Independent Consultant
Henrik Johanning
CEO, Senior Consultant, Genau & More A/S
Marina Levina
Product Owner-OSD, TTC- Tablets Technology Cell, GMS
GlaxoSmithKline
Roberto Margarita
Platform Director Corden Pharma
Luigi G. Martini
Chair of Pharmaceutical Innovation
King’s College London
Thomas Menzel
Menzel Fluid Solutions AG
Jim Miller
President,PharmSource Information Services
Colin Minchom
Senior Director Pharmaceutical Sciences Shire Pharmaceuticals
Clifford S. Mintz
President and Founder BioInsights
Tim Peterson
Transdermal Product Development Leader, Drug Delivery Systems Division, 3M
John Pritchard
Technical Director Philips Respironics
Thomas Rades
Professor, Research Chair in Formulation Desgin and Drug De-livery, University of Copenhagen
Rodolfo Romañach
Professor of Chemistry University of Puerto Rico, Puerto Rico
University of Ljubljana, Slovenia
Griet Van Vaerenbergh
Above is a partial list of the Pharmaceutical Technology brand editorial advisory members. The full board, which includes advisory members of Pharmaceutical Technology North America, can be found online at www.PharmTech.com/EAB. Pharmaceutical Technology publishes contributed technical articles that undergo a rigorous, double-blind peer-review process involving members of our distin-guished Editorial Advisory Board. Manuscripts for editorial consideration should be sent directly to Susan Haigney, managing editor, [email protected]. 10% Post
Consumer Waste Editorial: All submissions will be handled with reasonable care, but the publisher assumes no responsibility for safety of artwork, photographs, or manuscripts. Every precaution is taken to ensure accuracy, but the publisher cannot accept responsibility for the accuracy of information supplied herein or for any opinion expressed.
Subscriptions:
Pharmaceutical Technology Europe is free to qualiied subscribers in Europe.
To apply for a free subscription, or to change your name or address, go to PharmTech.com, click on Subscribe, & follow the prompts.
To cancel your subscription or to order back issues, please email your request to [email protected], putting PTE in the subject line.
Please quote your subscription number if you have it.
List Rental: Contact Sarah Darcy; Tel. +44 1244 629 326 Fax +44 1244 659 321
Reprints: Reprints of all articles in this issue and past issues are available (500 minimum).
Contact Brian Kolb at Wright’s Media, 2407 Timberloch Place, The Woodlands, TX 77380. Telephone: 877-652-5295 ext. 121. Email: [email protected].
Copyright 2016. Advanstar Communications (UK) Ltd. All rights reserved.
No part of this publication may be reproduced in any material form (including photocopying or storing it in any medium by electronic means and whether or not transiently or incidentally to some other use of this publication) without the written permission of the copyright owner except in accordance with the provisions of the Copyright, Designs & Patents Act (UK) 1988 or under the terms of a licence issued by the Copyright Licensing Agency, 90 Tottenham Court Road, London W1P 0LP, UK.
T H E F U T U R E I N I N J E C T A B L E S
The Parenteral Drug Association presents:
1
st
PDA Europe
Annual Meeting
201
6
30 June - 1 July
Root Cause
Investigation
30 June - 1 July
Development of a
Pre-Filled Syringe
30 June
Test Methods for
Pre-Filled Syringes
30 June
Cleaning and
Disinfection
30 June
How to Find the Right
GMP for APIs
28-29 June 2016
Estrel Hotel Berlin
Berlin | Germany
EDITOR’S COMMENT
The Repercussions of
Data Integrity Violations
P
harma has huge responsibilities resting on its shoulders to deliver safe and effective medicine. As the push for transparency heightens, the industry finds itself increasingly being scrutinized in many areas, from drug pricing and the reporting of all clinical trial results to business ethics, sales and marketing activities, and quality control of manufacturing operations. But how well is Pharma managing regulatory compliance? A PwC report (1) in March 2016 noted a rise in the number of pharmaceutical companies being issued warning letters by the United States Food and Drug Administration for GMP violations related to data integrity deficiencies. The three most common data integrity issues cited by FDA for inspections from 2013 to 2015 were the lack of controls to prevent alterations of data by staff, failure to maintain records of accurate data, and delayed reporting of data.Data integrity is fundamental in pharmaceutical manufacturing. It enables traceability of a batch to its origin and, more importantly, ensures that drugs are
made and tested according to the required quality standards. It is, therefore, crucial that data records are accurate, complete, legible, attributable, intact, and maintained within their original context, including their relationship to secondary data records. Non-compliance does not only result in revenue loss for the violator due to facility shutdown, product recalls, import bans, and delayed or denied drug approvals, but it also tarnishes the company’s reputation and provides competitors with an opportunity to increase their market share. In addition, warning letters divert the attention of workers from their daily activities to corrective and preventive actions, which cost significant time and money.
According to Ernst & Young (2), one the key root causes of data integrity issues is staff shortage combined with excessive workload that lead to inaccurate and incomplete documentation; the emphasis on quantity over quality forces workers to compromise on quality so that production targets and dispatch timelines can be met. The lack of awareness among employees and ineffective training are also known to result in data integrity problems.
Regulators worldwide are increasingly focusing on data integrity in their
enforcement efforts. Despite recent reports
of Indian and Chinese companies receiving warning letters for data integrity violations, the problem is, in reality, a global one. The solution for the industry does not only involve changing its approach from reactive to proactive but embracing a culture of quality so that regulatory compliance becomes second nature in every company.
References
1. PwC, Health Research Institute Regulatory Spotlight, Data Integrity Problems A Growing Risk to Global Pharma Companies (March 2016), http://pwchealth.com/cgi-local/hreg-ister.cgi/reg/pwc-data-integrity-spotlight.pdf, accessed 1 Apr. 2016.
2. Ernst & Young, Analyzing the State of Data Integrity Compliance in the Indian Pharmaceutical Industry (June 2015), www.ey.com/Publication/vwLUAssets/ ey-data-integrity-compliance-in-the- pharma-industry/$FILE/ey-data-integrity-compliance-in-the-pharma-industry.pdf, accessed 1 Apr. 2016.
Adeline Siew, PhD
Editor of Pharmaceutical Technology Europe
www.sterile.com
1-888-4-STERILE
For more than 30 years, VAI has pioneered the design
and manufacture of hundreds of clean room solutions.
Quadruple Bagged using the ABCD Introduction System®
t
Cleanest wipe
in the industry
t
Asepti-Fill® closed
i lling system
t
Laundered in Class 1
t
Saturated wipes
are made with WFI
t
Lot Specii c
Documention
for all wipers
t
Laser cut sealed edges
No other company of ers this broad a range of wipers…
DAS-Wipe®
Stainless Steel
cleaning and
lubricant wipe
STERI-PEROX®
WIPE
Saturated
Hydrogen
Peroxide Wipe
HYPO-CHLOR®
Wipe
Saturated Sodium
Hypochlorite Wipe
DEC-Clean®
Wipe
Removes residue
from disinfecting
agents
ALCOH-Wipe®
Saturated with
DECON-AHOL®
WFI 70% USP
Isopropyl Alcohol
STEEL-BRIGHT®
Wipe
Stainless Steel
Cleaning wipe
70% USP IPA
OUTSOURCING REVIEW
Jim Miller is president of PharmSource Information Services, Inc., and publisher of Bio/Pharmaceutical Outsourcing Report, tel. 703.383.4903, Twitter@JimPharmSource, [email protected], www.pharmsource.com.
E
arly development services are not the most glamorous segment of the contract development and manufacturing organization (CDMO) industry, but these days they may be the hottest. Booming market demand is driving record growth, expansion, and acquisition activity.Formulation development and manufacturing of early-phase clinical trial materials (CTM) historically have been some of the least attractive segments of the contract services industry from a business standpoint. Typically, price points were low relative to larger-scale manufacturing services, and client churn was high because projects were generally one-off engagements for emerging bio/pharma companies with short pipelines and limited funding. Further, the business had low barriers to entry and there were few economies of scale, so the industry remained highly fragmented.
Market and technical trends in recent years have drastically changed the significance and attractiveness of early development services. The serious formulation work is done in Phase II after proof-of-concept is demonstrated, and at that point, scale up and tech transfer to the commercial manufacturer is a much greater consideration. Commercial-scale contract manufacturing organizations (CMOs) have a strong interest in capturing the client at that point because they have the opportunity to retain the business as the product works through the development stages to commercialization. Clients have an interest in having the commercial CMO develop the formulation because they expect it will facilitate tech transfer with minimal need to change the formulation as it is scaled up.
Formulation development services
Formulation development has also taken on greater significance as delivery challenges have increased. For instance, today’s highly potent APIs often present major challenges for solubility and bioavailability that require more sophisticated formulations such as amorphous solid dispersions produced with advanced technologies such as spray drying or hot-melt extrusion. Advanced expertise is needed to select the most appropriate formulation and processing technology. Specialized knowhow mayalso be required as bio/pharma companies seek novel presentations that can differentiate their products in the market (e.g., taste-masked or controlled-release formulations).
Development services have also been beneficiaries of the boost in drug development activity fed by record amounts of external fundraising. Total non-debt fundraising increased by approximately 50% to US$31.5 billion (€27.68 billion), compared to US$21.5 billion (€18.9 billion) in 2014, according to data from the PharmSource Lead Sheet. With ample funding, clinical trial activity has been on the upswing. According to data from clinicaltrials.gov, Phase I and Phase II trial registrations declined from 2008 through 2013 when external funding was difficult to obtain, but registrations have been trending upward in the past two years.
Company investments
The increased demand for development services is showing up in the financial results of the few CDMOs that reveal them publicly. Catalent, Patheon, and the Metrics Contract Services unit of Mayne Pharma have all reported revenue growth of 20% or more for their development services businesses in the past year. Anecdotally, CDMOs report record levels of business and tight capacity that in many cases is forcing clients to wait six months for a project slot.
Not surprisingly the demand for formulation and other development services is driving both capacity expansions and new investor interest. For instance, Patheon has established a new development services unit at its Greenville, North Carolina, US, facility, while Catalent invested in its Kansas City, Missouri, US, facility as its centre for solid-dose development. Injectables capacity is a particular focus, owing to the growth of biologics: Alcami has announced expanded capacity at its Charleston, South Carolina, US, injectables operation, while Piramal Pharma Solutions has announced a US$10 million (€8.79 million) expansion of its Coldstream Laboratories facility in Lexington, Kentucky, US.
CMC Development is Hot
Demand is driving expansion and consolidation of formulation and clinical trial materials services.
OUTSOURCING REVIEW
Lonza’s entry into the market is strong confirmation of the opportunity for formulation and other development services. The company will launch formulation and analytical development in the fourth quarter of 2016; preclinical and GMP-compliant clinical supplies manufacturing will begin in 2017. The new drug product services offering will focus on parenteral dosage forms and will include specialized analytical services, such as particulate identification, characterization, extractables and leachables, and container/ closure integrity testing.
Adding clinical-scale drug product development and manufacture gives Lonza a more complete offering to help customers get into the clinic and makes the company more competitive with other biologics CDMOs. Its strategy for the business is to lead with strong technical capabilities, which will distinguish it from those fill/finish CDMOs that have no or limited pre-formulation and formulation capabilities.
Acquisition activity has picked up in the development services space in response to improved demand. At the end of 2015, Capsugel acquired Xcelience (Tampa, Florida, US) and its Powdersize micronization unit (Quakertown, Pennsylvania, US). They have become part of Capsugel’s Dosage Form Solutions (DFS) business unit, which includes Bend Research (Bend, Oregon, US) and Encap Drug Delivery (Livingston, UK).
The acquisition builds out the Capsugel DFS by adding micronization to DFS’s portfolio of solubility- and bioavailability-enhancing technologies, which already include spray drying, hot-melt extrusion, liquid-filled hard capsules, and softgels. Further, it bolsters Capsugel’s capabilities and capacity in analytical testing, formulation, and CTM manufacture of solid-dose products.
Other recent deals in the early development arena include AMRI’s acquisition of Whitehouse Laboratories, a provider of analytical testing services; and Amatsigroup’s (Fontenilles, France) acquisition of Q Biologicals (Zwijnaarde, Belgium). Amatsigroup is a private equity-backed French CDMO that has rolled up some smaller operations in France and Belgium. It is under the radar of most US-based companies but has now reached revenues of more than US$33 million (€30 million), which is decent for a standalone development services company.
One situation worth watching is whether preclinical contract research organization (CRO) Charles River Laboratories decides to ramp up its presence in the pharmaceutics arena. Charles River, which has revenues of US$1.4 billion (€1.23 million), recently announced plans to acquire WIL Research, whose QS Pharma unit provides formulation, analytical, and CTM manufacturing with revenues of more than US$50 million (€43.94 million) for those businesses. Charles River already has a major position in analytical testing for large-molecule analytical testing and CEO James Foster indicated at January’s JP Morgan Healthcare Conference that CMC services could be a new growth avenue for Charles River.
So the formulation and CTM services space is very much in play as its participants experience high double-digit growth. There are only a few independent service providers left in this segment of the industry so there aren’t many significant deals left to be done, but it is a part of the industry that is rapidly maturing. PTE
31
ST
INTERNATIONAL EXHIBITION FOR
FINE AND SPECIALITY CHEMICALS
www.
chemspeceurope
.com
1- 2 JUNE 2016
BASEL MESSE, SWITZERLAND
Royal Society of Chemistry Symposium
REACHReady Regulatory Services Conference
Agrochemical Intermediates Conference
The Pharma Outsourcing Best Practices Panel
TOP-CLASS CONFERENCES & WORKSHOPS!
OrganisersMackBrooksExhibitiond
EXHIBITION
CONFERENCE
NETWORKING
Chemspec Europe is exclusively dedicated to the fine, custom
and speciality chemicals sector. With its focussed profile it is
the
place to be for international industry professionals. Leading
manufacturers, suppliers and distributors will present a wide
spectrum of fine and speciality chemicals for various industry
sectors.
Sean Milmo
is a freelance writer based in Essex, UK, [email protected].
GL
OB
E
: Z
OON
A
R
R
F
/G
ET
T
Y
I
M
A
G
E
S
T
he European Commission (EC) has finally published a regula-tion on mandatory safety features for medicines packaging to combat counterfeiting of pharmaceuticals. Publication of the regulation means that a deadline of 9 Feb., 2019 has been set for implementation of European Union (EU) rules on unique identifier barcodes and anti-tamper devices for single-pack medicines, many years after the rules were first suggested. The three-year transition period, however, has already been ques-tioned by information technology (IT) specialists as to whether it will be long enough to allow time for the proper testing of systems for authentication of the unique identification data.The packaging safety features scheme will be an end-to-end system under which the serialization and other data on each pack will be fed by packaging units into a repositories network so that it can be verified by a pharmacist with a scanner at the dispensing point.
The EC, the Brussels-based EU executive, formally approved the regulation in October 2015, after the European Parliament and the European Council, representing the EU’s 28 member states, delegated it with the task of drawing up detailed rules on the single-pack serialization codes and their verification. The packaging regulation is one part of the 2011 Falsified Medicines Directive (FMD) of the EU, which was based on legislative proposals first made by the EC in 2008. Sections of the FMD, including those covering packaging, were drawn up to strengthen rules first outlined in a 2001 directive on medicines.
The regulation, which mostly applies to prescription medicines, has taken so long to work out after many years of consultation and bring to the stage of implementation because of the complexity of serialization systems. Of the many pieces of EU legislation on medicines, the packaging regulation could turn out to be the one with the biggest impact on the European pharmaceutical sector because of the broad range of groups that it affects.
Medicine manufacturers and packaging contractors, wholesalers and other distributors, and hospit al and communit y pharmacists all have to be linked to a new digitalized supply-chain system based on a network of data repositories containing the serialization and other data on each individual pack. In Germany alone, Europe’s largest pharmaceuticals market, there are approximately 20,000 pharmacists to be connected to the system.
The difficulties of setting up the safety features scheme within three years stem from the necessity to establish it on two levels—one on the manufacturing side and the other within a repositories network.
Implications for manufacturers
The manufacturers or their packaging contractors are having to reorganize and upgrade their packaging lines for the pro-duction of packs which, under the regulation, will be required to have a 2D matrix serialization barcode containing a product code, serial and batch numbers, and expiry date. The pack will also have to have an anti-tamper device (ATD), the speci-fications for which are not laid down by the regulation, except that it should not interfere with the verification process at the dispensing point. In most cases, manufacturers and pack-aging companies will have to rely on the outside expertise of software and equipment vendors, the most efficient of which already have full order books.
A survey in 2015 by Domino Printing Sciences, Cambridge, England, a specialist in packaging coding systems, found that approximately 35% of companies investing in packaging improvements to comply with the regulation were upgrading 100 or more packaging lines. Another 15% were converting
between 50 and 99 lines (1).
Around two-thirds of companies had allocated serialization budgets of more than £2 million (US$3 million). However, 25% had not fixed a budget, while around a quarter were also expecting that preparation for serialization would taken them longer than three years, according to the survey.
“A high number of smaller companies are still not even aware of the regulation so it is likely a lot of them will not make the deadline,” says Craig Stobie, head of Domino’s global life-sciences team. “On the other hand many of the larger and more proactive companies have been preparing for the regulation for some time.”
Creating a repositories network
Parallel with the preparation work being carried out by the manufacturers and their packaging partners is the creation of a repositories network. This network will be run by National Medicines Verification Organizations (NMVOs) in each of the EU’s 28 member states and a central European Medicines Verification Organization (EMVO) with a European Hub con-necting all the national systems to ensure their interoperability. The Hub will link the pharmacists authenticating the data on packs with the national schemes.
Under the regulation, authentication is the responsibility o f m a n u f a c t u re r s, dis t r ib u t o r s, a n d p h a r m a cis t s s o the verification organizations are being formed by the stakeholders, with much of the funding being provided by the manufacturers. The NMVOs will be supervised by national medicines authorities.
Europe Moves Forward on
Anticounterfeiting Measures
Pharmaceutical Technology Europe
APRIL 2016 11 The EMVO and the Hub has alreadybeen set up. Of the EU member states, only Germany has so far established an NMVO, but its authentication system is still in the pilot stage. Most of the rest are well advanced in the process of creating verification organizations so that they should be ready to sign contracts with authentication software providers later this year.
“It will be challenging (set ting up NMVOs on time), and for some countries it will be more challenging than others,” says Anci Kvarnstroem, a consultant at PhQConsulting in Sweden. “But they will all be able to meet the deadline. A lot of countries had already started preparing to implement the regulation even before it was finally published.”
Testing the system
“The most challenging task will not be the establishment of the NMVOs but putting in place and testing the actual authenti-cation systems in each country,” she con-tinued. “[These will have to have] secure connections to the European Hub and to all stakeholders that are obliged to verify and decommission the individual packs by the deadline in February 2019.”
T h e re is a g row in g v i ew a m o n g IT experts and groups involved in the creation of the NMVOs that a key issue is the time needed to test new systems. T hu s b ot h t h e m a nu f a c t u re r s a n d packaging companies and the NMVOs will have to allow for a sufficiently long testing period before the deadline of February 2019.
“This is a huge change-management project,” said Graham Smith, commercial d i r e c t o r a t A e g a t e , a U K- b a s e d a u t h e n t i c a t i o n s e r v i c e s p r o v i d e r. “Before the regulation comes into
effect in February 2019, we need to make sure everything works properly. Manufacturers need not only to serialize medicines, but also to test that these codes can be loaded into the database and that the codes can be read by wholesalers and pharmacists. It seems to make sense that once an authentication system is in place there should be sufficient time allowed for testing.”
Some stakeholder groups are aiming to leave as long as 18 months for testing by set ting up NMVOs as quickly as
possible and appointing authentication s e r v i c e p r o v i d e r s t o e s t a b l i s h verification systems by the end of 2016. In the UK, for example, stakeholders are expecting to have a verification system at least at the pilot stage by mid-2017 so that manufacturers will then be able to start testing their serialization systems on it.
“We believe that the testing period needs to be a lengthy one,” says Rick Greville, responsible for the implementa-tion of FMD legislaimplementa-tion at the Associaimplementa-tion of the British Pharmaceutical Industry (ABPI), representing UK manufacturers. “We are advising manufacturers that they should allow a minimum of six months before the February 2019 deadline for testing,” he says.
Experts have warned that manufac-turers need to ensure that all their lines upgraded for serialization are properly tested. “The temptation is to concen-trate on testing the better equipped lines while it is the lines held in reserve which will be causing problems,” says Stobie.
Opinion is nonetheless divided about the amount and application of the data provided by the new safety features system. The Brussels-based Medicines for Europe—formerly European Generic and Biosimilar medicines Association (EGA)—is, for example, urging medicines agencies to allow the application of the unique identifier data to be broadened to applications like pharmacovigilance. “The FMD legislation allows national competent authorities to extend the scope of the safet y features,” says Maarten Van Baelen, the association’s market access director.
However, others believe that the priority should be ensuring that the serialization system as laid down by the regulation is operating by the February 2019 deadline. “The potential for other data management
s c h e m e s i s v e r y a l l u r i n g ,” s a y s Kvarnstroem. “We must remain focused on the implementation of the serialization system to comply with the regulation. If we start to widen the scope of the project, we won’t be able to get it ready on time.”
Reference
1. C. Stobie. “Compliance Waits for No One!,” presentation at Pharmapack con-ference (Paris, February 2016). PTE
THE
SCIENCE OF
COMPLIANCE
[email protected]
www.starna.com
+44(0) 20 8501 5550
The World Leader in UV,
9
LVLEOH
DQG1
,
5&
HUWL
Ć
H
G
Lifetime
G
uarantee
Fast Recalibration
Service
4001
REFERENCE MATERIALS
GL
OB
E
: Z
OON
A
R
R
F
/G
ET
T
Y
I
M
A
G
E
S
T
he Ebola outbreak in 2015 and the current stampede to develop a new vaccine to combat the Zika virus illustrate the increas-ingly important role of vaccines in advancing public health. Public and private funding for vaccine development and production has grown noticeably, from US$822 million (€722 million) in 2000 to US$3.6 billion (€3.16 billion) in 2014, spurred by the “Decade of Vaccines” (2011–2020) initiative launched by the Bill & Melinda Gates Foundation and the global health community five years ago. The campaign has helped move more vaccine candidates through clinical testing to market approval, according to an analysis in the February 2016 special issue on vaccines in the journal Health Affairs (1). And it has prompted manufacturers to expand and mod-ernize vaccine production facilities to be able to meet the global demand for treatments and to avoid product shortages.The urgent need for new treatments to combat Zika and related birth defects also illustrates the inadequacies in the current world health infrastructure to combat epidemic and pandemic diseases. Experts seek to improve public health infrastructure and to reach global agreement on clinical trial design and regulator y decision making to support rapid development of needed therapies. At a Ministerial Conference on Immunization in Africa in February 2016, officials emphasized the need for greater funding of national immunization programmes, including expanded regional capacity for vaccine development and production; they also demanded increased price transparency to facilitate access to affordable vaccines.
Scientific advances
To achieve these goals, scientists in academia, government, and industry are moving to tap new genomics technology to reduce the long R&D process and help modernize vaccine development and production. The design of new antigens based on molecular structure and advances in synthetic biology will help deliver new vaccines to patients, noted GlaxoSmithKline (GSK) Chief Scientist Rino Rappuoli at the PDA/FDA Vaccines conference in December 2015.
Such strategies may accelerate development of a vaccine for Zika, which is similar to that for dengue, East Nile virus, and yellow fever. Sanofi hopes to build on its success in producing a vaccine against dengue fever, which was approved in December 2015 in Mexico, Brazil, and the Philippines (2). GSK, Pfizer, Merck, Johnson & Johnson, and Takeda, as well as several smaller biotech firms, are examining vaccine research portfolios for potential candidates to combat Zika.
Anthony Fauci, director of the National Institute of Allergy and Infectious Diseases (NIAID), has explained at several Congressional hearings that he hopes to begin Phase I studies
on a Zika virus vaccine in a few months, building on a DNA-based vaccine for West Nile virus already under development. Positive early results could lead to larger trials more quickly than usual, but a fully tested and approved Zika vaccine still will take several years.
Meanwhile, testing continues on Ebola vaccine candidates that have produced evidence of safety and efficacy from “emergency use” and initial clinical studies. Merck announced advanced clinical trials for its vaccine developed with NewLink Genetics in January 2016, plus a deal with GAVI, the Vaccine Alliance, for coverage and distribution by the end of 2017 (3).
Vaccines for other widespread infectious diseases also are moving forward. Researchers recently reported progress in combating Middle East Respiratory Syndrome (MERS), and US investment in research to protect against bioterrorism has led to a new vaccine to treat anthrax exposure. Efforts to establish more reliable and efficient supplies of seasonal influenza vaccine in the United States, moreover, have led to development and US Food and Drug Administration (FDA) approval of the first US adjuvant flu vaccine.
President Obama has asked Congress for US$1.9 billion (€1.67 billion) in emergency funding to tackle Zika, much of that designated for the Centers for Disease Control and Prevention (CDC) to track the disease and prevent its spread, and to the National Institutes of Health (NIH) to fund R&D on vaccines and antiviral therapeutics. FDA would get US$10 million (€8.78 million) to support oversight of vaccines and diagnostics, as well as assays to screen blood for the Zika virus. Additional resources would help the US Department of Health and Human Services’ (HHS) Biomedical Advanced Research and Development Authority (BARDA) further the development and manufacturing of new Zika vaccine candidates and other innovative vaccine platform technologies. BARDA already is
Vaccine Development Faces
Urgency and Challenges
Global outbreaks energize vaccine R&D and drive production modernization.
Jill Wechsler is Pharmaceutical Technology Europe’s Washington editor, tel. +1 301.656.4634, [email protected].
Scientists are moving
to tap new genomics
technology to reduce the
long R&D process and
help modernize vaccine
Pharmaceutical Technology Europe
APRIL 2016 13 working with Brazilian agencies todevelop a Zika vaccine and to build commercial-scale manufacturing.
Manufacturing challenges
In addition to support for innovation to achieve a sustainable and profitable vac-cine industry, manufacturers hope to gain more streamlined vaccine development requirements and clearer reimbursement and coverage programmes. Despite the proven effectiveness and value of vac-cines, health agencies and insurers want lower prices, while conflicting regulations add to manufacturing costs.
F o r e x a m p l e , t h e t r a n s f e r o f production of a long-used vaccine adjuvant to a new facility at GSK’s Marburg, Germany, production site took five years and involved 15,000 pages of nearly 300 documents, reported GSK officials Hans-Joachim Mai and Jutta Ochs at the 2015 PDA vaccines conference. The extensive process was needed to document unchanged quality attributes of the adjuvant under a new automated quality control system.
Similarly, Wilson Forsyth, director of quality control at AstraZeneca (AZ) Biologics Global Operations, described a complex two-year process for shifting a vaccine quality control operation from California to the UK as part of a consolidation of quality control and vaccine R&D at AZ’s Liverpool facility. The transfer process involved multiple work streams and was complicated by different time zones and the need to continue production of seasonal influenza vaccine.
More modern vaccine production aims to avoid supply shortages, which regularly plague flu vaccine makers due to tight production timelines. In 2015, AZ’s MedImmune experienced production “challenges” that delayed shipment of millions of doses to the US of its FluMist nasal spray. Vaccine shortages are difficult to resolve because the cost of building and operating high-quality vaccine production facilities limits availability of alternate sources and the ability to outsource production.
Costs and coverage
Vaccine shortages may be most common with low-cost products because
manu-facturers are more likely to close a plant that experiences vaccine quality produc-tion issues or to exit that market rather than invest in improvements, according to a recent analysis of the relationship between vaccine prices and shortages by David Ridley and colleagues at the Duke University Fuqua School of Business (4). The analysts found that most vaccine
shortages involved older, low-cost prod-ucts, and fewer problems with newer products, particularly innovative combi-nation vaccines. Government health pro-grammes and other payers want to avoid over-paying for vaccines, Ridley noted, but higher-priced products encourage R&D in new vaccines and in high-quality and high-capacity manufacturing facilities.
Co-pays and coverage limits by insurers also can block access to vaccines, as seen with Medicare beneficiaries who have to pay more out-of-pocket for vaccines, according to a recent study by Avalere Health for GSK (5).
References
1. Health Affairs 35 (2) (February 2016), http:// content.healthaffairs.org/content/35/2.toc 2. Sanofi Pasteur, “Sanofi Pasteur’s Dengue
Vaccine Approved in the Philippines,” Press Release, 22 Dec., 2015, http://hugin. info/152918/R/1975412/722908.pdf 3. Gavi, “Ebola vaccine purchasing
commit-ment from Gavi to prepare for future out-breaks,” Press Release, 20 Jan., 2016, www. gavi.org/Library/News/Press-releases/2016/ Ebola-vaccine-purchasing-commitment- from-Gavi-to-prepare-for-future-out-breaks/
4. D. Ridley, X. Bei, E. Liebman, Health Af-fairs 35 (2) (February 2016), http://content. healthaffairs.org/content/35/2/235.abstract 5. Avalere, “Medicare Has the Potential to
Avoid Preventable Illnesses by Encourag-ing Broader Coverage for Adult Vaccines,” Press Release, 18 Feb., 2016, http://avalere- health-production.s3.amazonaws.com/up-loads/pdfs/1455800241_20160217_Part_D_ vaccine_coverage_release.pdf PTE
Find Out More
:
website
- www.mournetrainingservices.co.uk
- [email protected]
telephone
- +44 28 3083 4938
Validation & Transfer of Methods
for Pharmaceutical Analysis
External Course Dates in 2016:
London
- 18
thto 20
thMay & 19
thto 21
stOctober
Dublin
- 15
thto 17
thJune
Berlin
- 28
thto 30
thNovember
This 3 day training course, designed and delivered by our analytical
sciences guru Mrs Oona McPolin BSc, MSc, CSci, CChem, MRSC
-will explain everything that you need to know about validating and
transferring methods, including compliance with ICH guidance and
Re
so
lu
tio
n
P
ro
duc
tio
ns
/G
et
ty
Image
s
ime travelers from the 1980s would not recognize the typical pharmaceutical laboratory of 2016. Large spaces filled with dedicated tabletop instruments have disappeared, as pharmaceutical companies have shut down facilities, downsized, and are outsourcing more analytical laboratory work to contract analytical service providers.
As pharmaceutical facilities become modular and more flexible, instruments are shrinking. Portable devices bring more data to the warehouse, plant floor, and the lab, while wireless operation and use of radiofrequency identification (RFID) tags are being used to reduce operator error.
Data manipulation has been simplified in more user-friendly chemometrics and statistical analysis software. In addition, procedures such as chromatography have become easier and more powerful, while workhorse methods are gaining ground as “first-pass” alternatives to expensive and complex analytical methods.
To wrest value from every procedure and instrument, laboratories are embracing some of the basic concepts of lean manufacturing and optimizing workflow to reduce waste and improve efficiency. “With a move toward outsourcing and greater focus on biotherapeutics, customers are asking for more and better workflow solutions,” says John Rontree, senior director, chromatography and mass spectrometry at Thermo Fisher Scientific. “These solutions simplify tasks for outsourcing vendors, and reduce the complexity that comes with analyzing biologics,” he says.
A growing number of pharmaceutical companies are outsourcing most, if not all of their analytical work. Today, stability testing, dissolution, and method development are typically outsourced, says Keith Moore, vice-president of analytical services at Metrics Contract Services.
Standard processes include high-performance liquid chromatography (HPLC) assays, dissolution testing by either HPLC or ultra violet (UV), and Karl Fischer testing procedures, says Moore. Pharmaceutical manufacturers are also outsourcing operations that require specialized equipment and highly trained users, such as mass spectrometry, X-ray powder diffraction (XRPD), and nuclear magnetic resonance (NMR) spectroscopy, he says.
Pharmaceutical manufacturers either outsource directly to contract laboratory companies or to larger contract manufacturing organizations (CMOs) that have acquired analytical capabilities. Metrics, for example, is a contract development and manufacturing organization (CDMO) with analytical testing capabilities.
According to Industry Standard Research’s reports on the state of pharma outsourcing, lab services are the seventh most widely outsourced function for small molecules and the sixth largest for biopharmaceuticals. It’s also an area that is seeing more mergers and acquisitions. Eurofins, for instance, which bought Lancaster Labs in 2011, acquired Sinensis Life Sciences, an analytical services company in the Netherlands, in 2016. At the end of December Agnes Shanley
Advances for
Bio/Pharma
Analytical
Laboratories
Lab Procedures
2015, AMRI bought Whitehouse Analytical, a major analytical services company.
Both pharmaceutical sponsors and their contract partners benefit from more powerful analytical methods. The workhorse method, HPLC, still dominates pharma labs, but alternatives such as UHPLC (ultra-high pressure liquid chromatography), offered by leading chromatography vendors, including Waters, Varian, Perkin Elmer, and Thermo Scientific, are gaining users.
UHPLC, which uses smaller-diameter resin particles to improve resolution, speed, and sensitivity, allows more samples to be analyzed faster than with other methods. In addition, it reduces solvent requirements and waste for disposal, and requires less space, hood space, and storage.
UHPLC for analytics has been likened to continuous processing in the manufacturing world. “If continuous processing reduces the cost of goods sold, UHPLC lowers the cost of analysis,” notes Emil Ciurczak, NIR spectroscopist who published the industry’s first papers on the use of process analytical technology (PAT) in pharmaceutical manufacturing when he worked at Sandoz in the 1980s.
“With UHPLC, less material is required, and less time is needed for preparation,” Ciurczak says. “So, if, for example, you move sampling from 40 mLs to 1–2 mLs per run, you also save the cost of buying, storing, and disposing of materials,” he says. “The throughput increases are especially remarkable, with one instrument capable, in some cases, of doing the work of six conventional HPLCs,” he adds.
For bioseparations, notoriously difficult to achieve efficiently, vendors such as BIA Separations, Diosynth, Biorad, Agilent, and Merck Millipore have developed monolithic columns
that improve mass transfer and reduce purification times.
Continuous and simulated moving bed (SMB) chromatography, which allows several columns to be run in parallel, has also been developed by companies that include Novasep, GE Healthcare, Tarpon Biosystems, Knauer, ChromaCon, and Semba Biosciences. Vendors claim that this approach can improve productivity by a factor of up to six, and reduce the use of resin and buffer by 75% (1).
For dissolution testing, Moore says, UV spectroscopy is often the first choice, even before HPLC, because it provides immediate data for trending and saves solvent, disposal, and other costs. It also uses one value, absorbance, to analyze data, so there is no need to transfer samples to vials.
In some situations, Fourier-Transform infrared (FTIR) and Raman spectroscopy are being used to evaluate polymorphism, instead of the more challenging x-ray powder diffraction.
In general, chemical imaging is becoming more important, says Ciurczak, and vendors such as Innopharma, which developed a real-time particle size imaging platform with Glatt in 2015, as well as Chemimage, Malvern, Bruker, and Perkin-Elmer, are improving their offerings in this segment.
Use of ion mobility spectroscopy is also increasing for some applications. “It’s becoming the poor man’s mass spectrometer,” he says. Originally used in airport security applications, the technique, offered by vendors that include Smith’s Detection, Water, SelexION, and SCIEX, found its first pharma applications in cleaning validation back in the late 1990s. Today, it is finding increased use in some lower-end mass spectrometry
applications, and is also taking over some HPLC applications. “If you can get the equivalent of HPLC testing in 20 milliseconds without column or solvent, why not?” Ciurczak asks. Although the technique cannot be used for stability studies, he says, it’s becoming a handy quality control (QC) tool. “In some cases, it can be more specific than Raman or near infrared (NIR), and it’s also convenient for warehouse applications.”
Use of mass spectroscopy is also increasing, however, and benefitting by improved workflows and features that can streamline processes, eliminate errors, and improve access to data, says Rontree. “The technology may become more sophisticated, but a single system today can provide more information [than was possible in the past] and reduce the potential for errors,” he adds.
Increased outsourcing has changed the equation for laboratory data access and manipulation. Laboratory information management systems (LIMS) have become data storage systems, from which windows can be opened to allow access to and use of data. Some contract labs offer clients access to relevant portions of their LIMS in systems such as Eurofins Lancaster’s LabAccess. In other cases, contractors use a portion of their client’s IT platforms or install required data-access systems.
Instrumentation is also being developed so that contract partners can have immediate access to data. “Certainly the cloud is playing an increasingly important role as customers look to personalization and access to private information,” says Rontree.
“There is also growing demand for more reliable approaches to method transfer,” he adds, which the company aimed to address with its AppsLab product, which allows methods to be accessed online and transferred directly without user intervention.
Another enhancement has been availability of simplified chemometrics packages. As Ciurczak notes, these
Lab Procedures
can save on training costs, and also mean that every location needn’t invest $10,000 in software licenses. In a typical pharma setup today, he says, software resides at the sponsor company’s home base, but results are cached so that it can be used at a contract partner’s facility or in the field.
In today’s downsized environment, more pharmaceutical manufacturers are taking lean manufacturing approaches, developed for the plant floor, to the laboratory. The idea is to reduce waste, whether of time, space, or resources, to increase the value of each work step and procedure and improve productivity.
All analytical platform vendors are taking these needs into account with equipment and software that improves workflow. Mettler-Toledo has worked with the Swiss Lean consultant Irwin Studer to examine specific problems in the lab. The biggest problems, says Studer, include time wasted waiting for measurement results, too many working steps, interface problems, where production is held up waiting for lab results, and incorrectly labelled samples.
Mettler launched a Lean Labs platform in October 2015, which includes improved sample titration and weighing systems, as well as wireless data transmission and radiofrequency identification (RFID) readers and writers, to allow data to be picked up automatically from RFID tags on sample containers, pipettes, and dosing heads, reducing error (2).
Portable devices, including handheld Raman and NIR spectrometers, are nothing new in pharma. Spinoffs of technology originally developed for the telecommunications industry, miniaturized spectrometers and imaging devices began to appear more than 10 years ago, but the technology has been steadily improving, Ciurczak says.
Today, batteries are much better and hold charge much longer than
they did in the early 2000s, and the devices are getting smaller, he says. Some portable imaging systems from leading vendors are beginning to look more like the tricorders made popular by Star Trek, held in holsters, says Ciurczak. He imagines utility belts holding swappable heads for use with different materials, as well as holders for batteries, being worn by the QC technicians and analysts of the future.
The pharmaceutical industry is already developing new approaches to buildings that would eliminate storage space and maximize flexibility, such as the PCMM (portable, continuous, miniature, and modular) systems developed by GEA Pharma Systems and G-CON Manufacturing, and evaluated by Pfizer and GlaxoSmithKline for oral solid-dosage forms manufacturing. This concept conjures images of the mobile QC technician being deployed throughout the facility, using technology that can move, seamlessly, from lab to process.
Despite the availability of portable, miniature instruments, this is still far from reality at most pharmaceutical plants. It may be coming closer, however, as portable NIR, Raman, and other imaging systems are used throughout the pharma facility.
Portability would represent a major change for an industry that has always kept lab and plant staff separate. Ciurczak recalls working for a pharmaceutical company and suggesting that lab technicians go out and insert NIR probes into the material being blended, instead of following the tedious but time-honored approach of sampling, delivering samples to the lab, and analyzing them by Karl Fischer and HPLC. There was no question of sending lab people to the plant, and, as a result, his team had to wait a week for answers, he says. Of course, union rules and restrictions were the first obstacle, Ciurczak says, but mindset might also play some part. “These are not technical problems but rather psychological ones,” says Ciurczak. “The equipment
for doing this may exist, but the will to use it doesn’t.”
However, Ciurczak sees change coming in work underway, for instance, at Rutgers University, which is working to standardize
ab initio concepts for continuous
manufacturing, focusing on powder flow and behaviour. Instruments used in this research will be calibrated in the lab and used throughout the facility, he says, essentially becoming both process and lab instruments and merging the two worlds.
There may be other reasons why quality and manufacturing are kept in such different worlds, such as the need to ensure that manufacturing goals do not influence (or potentially weaken) quality standards.
However, some of the industry’s strongest programmes in lean and continuous quality improvement, at facilities such as Wyeth’s in Pearl River (started before the Pfizer merger) have brought quality and manufacturing professionals closer together (3). Perhaps quality standards could be maintained, or even elevated, in a system where quality staff were closer to those performing the manufacturing work. But, whatever shape the pharmaceutical laboratory takes in the future, instrumentation, methods, and services are clearly changing along with the industry and the pharmaceutical facility.
1. Novasep, BioSC Continuous
Chromatography for Biologics, www.
novasep.com/technologies/biosc-con-tinuous-chromatography-for-biologics. html, accessed 28 Jan., 2016.
2. “Weighing Equipment Uses Lean
Laboratory Principles,” PharmTech. com, 13 Oct. 2015, www.pharmtech.
com/weighing-equipment-uses-lean-laboratory-principles, accessed 26
Jan., 2016.
3. 2007 Team of the Year Award:
Wyeth’s Pearl River biotech Fill-Finish Optimization Team,
Eliminate the ups and downs of continually replacing
and retraining temps by retaining our scientists at your
site. Hired, trained and managed by us, our
award-winning Professional Scientific Services
SM(PSS):
t
Eliminates headcount, co-employment and
project management worries
t
Avoids Temp turnover rate with managed insourcing
t
Costs you less than your own full-time employees
t
Delivers a 50-year history of regulatory compliant
technical expertise in your lab
t
Holds numerous client awards as the top
insourcing service provider for the past 10 years
Choose the PSS Insourcing solution that enables
us to keep staff grounded.
Tired of Your
Temps Bouncing?
Partner and prosper with our award-winning PSS.
Mari
a
T
o
utoudaki/
g
et
ty
i
mag
es
T
he compatibility of a biologic formulation with its primary packaging and container closure system is key to maintaining the stability of drug product and preserving its safety and efficacy. PharmaceuticalTechnology Europe spoke to subject matter experts
from One 2 One about the importance of choosing the right container and container closure system for biologics formulations and understanding the impact of primary packaging materials on the drug product. Hospira, One 2 One’s parent company, was acquired by Pfizer in September 2015. Experts include Lisa Cherry, R&D manager; Martin Gonzalez, PhD, senior group leader, R&D; and Shen Chen, PhD, director, Pharmaceutical R&D, all three from One 2 One Global Pharmaceutical Services; and Kerry Mulvaney, senior scientist, MS&T, Global Technology Services at Hospira, a Pfizer company.
Selecting the right container
PTE: Why is it important to select the right container and container closure system for biologic formulations?
One 2 One: Maintaining stability of a large molecule is of utmost importance and has to be considered at every step of the manufacturing and packaging process. Metal ions, pH, salts, silicone, gases, and leachables can all affect the stability in a container closure system. If a biomolecule becomes unstable, resulting in structural damage of the protein, it will lead to a propensity for aggregation of the drug product. Aggregation is a safety and efficacy issue that can lead to an immunogenic response within a patient. This means that the patient’s immune system could generate antibodies to attack the drug product, which
can lead to drug clearance or neutralization. The bottom line is that choosing the wrong container and container closure system may put patients in jeopardy, especially if they are taking medication for a serious condition.
PTE: What are the causes of incompatibility between a biologic formulation and its container?
One 2 One: Incompatibility can occur by not fully understanding the material surface properties of the container. Some characteristics to consider include the container surface chemistry, surface wettability, dimensional consistency, and surface roughness. Protein may also adsorb to silicone oil, which is often used in prefilled syringe and cartridge systems. In other words, we need to consider any material characteristics that may lead to protein adsorption because adsorption can lead to protein aggregation.
Another source for incompatibility would be with the rubber formulation for the stopper, plunger, or seal. Rubber components often contain additives and fillers to help strengthen the components. These additives and fillers, however, may contain trace metals that could leach into the drug product over time. This type of contamination may lead to protein damage.
PTE: Can you talk about the components of the primary packaging materials and their impact on a biologic formulation?
One 2 One: Typical containers for drug product biologics are made of glass or plastic and include vials, prefilled syringes, and cartridges. Each has a different set of closures: rubber stoppers for vials; plunger, needle, and needle shield (or luer tip and cap) for prefilled syringes; and plunger and seal for cartridges.
A Q&A by Adeline Siew, PhD
Container
Selection
for Biologic
Formulations
Formulation
All three container systems carry risk of interactions. We need to consider potential rubber-component leachables, as well as protein-silicone interactions. In particular, non-uniform silicone coating of containers is a real concern for protein stability over time. Tungsten may also be a problem. Prefilled syringes may be manufactured using a tungsten pin to shape the syringe tip. Residual tungsten in the syringe is a concern for biologics; it may become oxidized, cause a pH shift in the drug product solution, and lead to induction of protein aggregation. Prefilled syringe manufacturers are aware of this concern and are now marketing low-tungsten-containing syringes.
PTE: So what are the key considerations when selecting a container and container closure system for a biologic drug product?
One 2 One: The key consideration for initial container selection should be to choose components that maintain product stability by minimizing protein adsorption, extractables/ leachables, oxidation, and pH changes. Another is the intended route of administration. Container selection for a liquid medicine, for example, requires different considerations than a lyophilized one. For instance, evaluating protein interaction with silicone in a prefilled syringe or cartridge system is more important for a liquid than a lyophilized medicine. Components must also meet functional requirements to ensure safety at the point of administration and to protect the purity of the drug product throughout its shelf life. Finally, to expedite medications that meet unmet medical needs so that patients get them more quickly, choosing a reliable, tried-and-true container closure system such as a vial may facilitate market approval. More novel container closure systems could follow according to lifecycle management strategy.
PTE: Which is better for biologics, glass or plastic? How do you decide which to use?
One 2 One: This is really molecule- and formulation-dependent. Knowing the molecule’s sensitivities is imperative. For example, if a biomolecule is sensitive to oxygen,
a semi-permeable plastic container may not be optimal. However, if a biomolecule formulation leads to delamination in glass and does not exhibit oxygen sensitivity, a plastic container may be ideal. Certain types of glass and plastics can also cause wider variations in the drug product solution’s pH; again, pH changes can affect product stability, leading to potential protein aggregation.
In addition, we need to evaluate end-user requirements. A medication in one container closure system may be better suited for an emergency-care setting rather than for self-administration. Human-factor studies provide valuable information for assessing an appropriate container closure system from a patient usability perspective.
PTE: What testing or
characterization studies do you have to carry out to determine compatibility of the biologic formulation with the container and container closure system?
One 2 One: The main objective for determining compatibility would be to ensure that the biomolecule is stable in its container closure system. A typically ‘happy’ protein will exhibit maximum biological potency and minimal aggregation, denaturation, and/or damage. Initially, we can perform material contact studies to examine adsorption of the molecule to relevant container closure materials as well as contact effects on the potency and other critical quality attributes of the drug product. This type of study could be used for screening materials (for example, glass with different coatings) as well.
Once we choose a container closure system, filled drug product units should be tested under stress (temperature, humidity, light, and agitation) and normal storage conditions. For certain types of
containers, functionality testing is extremely important. Prefilled syringe and cartridge systems require proper functionality to deliver the intended dose. Glide force and break-loose force are common tests to ensure the liquid drug product is delivered smoothly and accurately. These types of mechanical tests should be included with stability studies.
PTE: What about the issue of extractables and leachables?
One 2 One: Rubber components are a significant contributor to leachables because they contain additives and fillers including transition metals, which could leach into the drug product over time. This type of contamination may lead to protein damage, especially when the product is exposed to agitation (air-liquid interface exposure). Another source of recent concern would be leachables from the adhesive used to attach the needle to a prefilled syringe system.
Stability testing of the drug product should monitor protein stability; changes in protein aggregation, oxidation, degradation, pH changes, and particulate matter can be indicators of compatibility. Stress studies, including agitation, can also provide key insights to compatibility.
Packaging trends
PTE: What recent trends do you see in primary packaging of biologic drugs?
One 2 One: As patients are taking a more active role in their healthcare, we are seeing more and more packaging systems that provide greater ease of use. Glass vial presentations have been moving towards prefilled syringe systems, especially as part of a lifecycle management approach. Dual-chamber prefilled syringes, allowing for mixing of a drug product immediately prior to application, have developed interesting applications in the biopharmaceutical market. There is some movement towards the use of layered plastic containers, such as cyclic olefin copolymer and cyclic olefin polymer, which prevent gas transmission into the drug product. Specialty pens, patches, and containers for self-infusion pump devices are also popular. PTE
“The main objective for
determining compatibility
would be to ensure that
the biomolecule is stable
in its container closure
system.”
—One 2 One
S
im
m
iS
im
o
n
s/
ge
tt
y im
a
g
e
s
L
yophilization, or freeze-drying, is a process used to stabilize a pharmaceutical formulation and increase the shelflife by removing water from the drug product. During lyophilization, the drug formulation is first frozen and then the ice is removed by sublimation under vacuum during a primary drying phase. A secondary drying phase is then used to remove unfrozen water molecules at a temperature higher than that used for primary drying. Pharmaceutical freeze drying cycles are designed to remove most of the loosely bound water and to achieve a pharmaceutically elegant cake. For biological materials, it is important to retain a high level of activity in the final product.Residual moisture content
Determining the residual moisture content of a lyophilized pharmaceutical product is important for several reasons. First, the amount of residual moisture content is related to the stability of the formulation over the shelflife of the product. Small-molecule formulations can have direct degradation pathways triggered by water, and it is crucial that all final product is below a defined residual moisture specification. In general, the degradation pathways for large-molecule formulations are more complex, with water often playing an indirect role. Second, moisture analysis of a statistically relevant sample set can give insight into the freeze-drying process itself. Residual moisture determination can be used as a tool in process studies to confirm the efficiency, consistency, and robustness of a specific freeze-drying cycle that has been designed for a particular drug formulation.
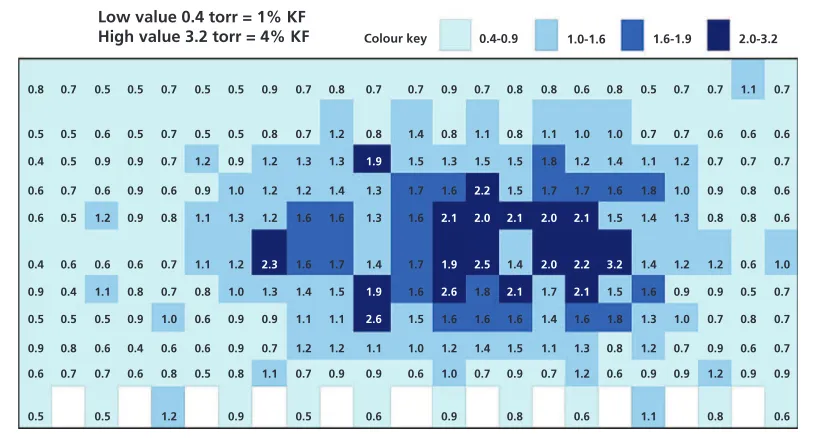
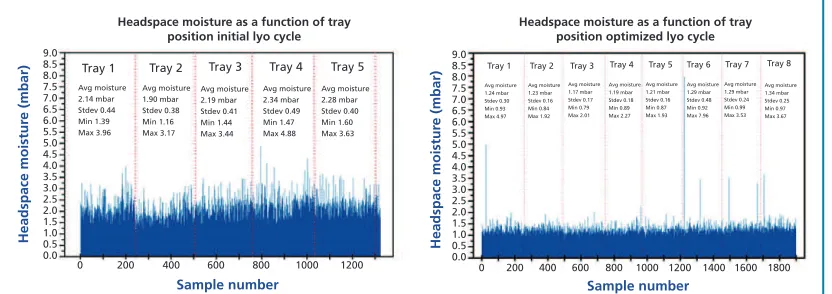
Typical pharmaceutical freeze-drying cycles usually target residual moisture contents in the range of 1% to 3% water by weight. Historically, a strategy that can be described as “the drier, the better” was often followed. For small molecules having a direct degradation pathway triggered by water, this approach was an appropriate strategy. However, in the world of large biopharmaceutical molecules, it is possible to over-dry. Studies have shown that even in the lyophilized state, proteins depend on small quantities of water to help maintain higher-order structure. Other types of products, such as certain lyophilized blood plasma formulations, need a minimum amount of water to achieve efficient dry-heat viral inactivation. It is therefore sometimes necessary to design a freeze-drying cycle that keeps all product vials within a certain moisture range, having both minimum and maximum specifications.
Historical cycles have often been too conservative (i.e., too long), meaning that the final product was over-dried, because research and development efforts did not take the time to optimize both the formulation and the freeze-drying cycle. Although conservative cycles produce product that meets quality parameters (i.e., sufficiently low residual moisture), the same product quality could be produced with much shorter cycles if appropriate studies are done. Current scientific approaches use various tools to monitor the lyophilization process and analyze the finished product with the goal of defining optimum freeze-drying cycles on a per formulation basis. Data are generated to demonstrate that product quality parameters (such as stability, cake appearance, appropriate reconstitution, and
Derek Duncan, PhD, is Director of Lighthouse Instruments, Science Park 408, 1098 XH Amsterdam, The Netherlands.