Involvement of potassium channels in the protective effect of
17
b
-estradiol on hypercholesterolemic rabbit carotid artery
Khadija Ghanam *, Leng Ea-Kim, James Javellaud, Nicole Oudart
Laboratoire de Pharmacologie,Faculte´ de Pharmacie,2 rue du Dr Marcland,87025 Limoges ce´dex, France
Received 1 June 1999; received in revised form 14 October 1999; accepted 2 November 1999
Abstract
The involvement of endothelium-derived hyperpolarizing factor (EDHF) in the protective effect of 17b-estradiol was investi-gated on the phenylephrine-precontracted carotid artery from cholesterol fed rabbits. Animals were fed for 8 weeks as follows: control group, standard chow; (control+estradiol) group, standard chow+17b-estradiol; standard chow+1% cholesterol, cholesterol group; or (cholesterol+estradiol) group, 1% cholesterol chow+17b-estradiol. Relaxations to acetylcholine (ACh) (3 nM – 30mM) were performed with Nv nitro-L-arginine methyl ester (300 mM) and indomethacin (10mM). Charybdotoxin (50 nM)+apamin (50 nM), glibenclamide (10 mM) or 4-aminopyridine (1 mM) were used to block, respectively, calcium-activated-K+, adenosine triphosphate (ATP)-sensitive-K+ and voltage-dependent K+ channels. In the control group, ACh induced a residual concentration-dependent relaxation. This response was impaired by hypercholesterolemia and restored by 17b-estradiol. In control and cholesterol groups, 4-aminopyridine or glibenclamide did not affect this relaxation, but in (control+estradiol) and (cholesterol+estradiol) groups, glibenclamide suppressed it. In all groups, this persisting relaxation was completely abolished by charybdotoxin alone or with apamin, by hemoglobin (10 mM), a nitric oxide scavenger, or by LY83183 (10 mM), a guanylate cyclase inhibitor. Thus, in the rabbit carotid artery, the protective effect of 17b-estradiol against hypercholesterolemia is probably mediated by a nitric oxide/cyclic GMP pathway which activates calcium-targeted and ATP-dependent K+ channels. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:17b-Estradiol; Nitric oxide; Hypercholesterolemia; Acetylcholine; Carotid artery; K+channels
www.elsevier.com/locate/atherosclerosis
1. Introduction
Several studies, in vitro and in vivo, have reported that endothelium-dependent relaxations are due essen-tially to release of three vasodilator factors: nitric oxide [1,2], prostacyclin [3] and endothelium-derived hyperpo-larizing factor (EDHF) [4 – 6]. Both nitric oxide and prostacyclin effects have been extensively investigated, while the nature and the site of action of EDHF remain unclear. It was proposed that EDHF induces smooth muscle relaxation by hyperpolarizing the membrane of vascular smooth muscle. The stimulation of this en-dothelium-dependent hyperpolarization is due to the opening of K+ channels [7,8]. Moreover, the nature of targeted K+ channels is variable, depending upon
spe-cies, arteries and agonists [5,9,10]. The identity of this factor was also controversial. It may be a product of the cytochrome P450 pathway [11], a cannabinoid [12], K+ [13] or nitric oxide [14 – 16].
Cardiovascular diseases such as hypercholesterolemia or atherosclerosis are associated with an impairment of endothelium-dependent relaxations [17,18]. It has been reported that this dysfunction is due to an alteration of nitric oxide synthase [19] or an increased degradation of nitric oxide by oxidants [20,21]. The contribution of EDHF under these pathophysiological conditions re-mains incompletely understood. Some reports have shown a reduction of EDHF-induced relaxations in a model of hypercholesterolemia [22,23].
Impairment of endothelium-dependent vasorelax-ation in hypercholesterolemia was prevented by chronic treatment with 17b-estradiol, via either an increase of nitric oxide synthesis on the vessel wall [24], its effect on plasma lipids [25], or the regulation of antioxidant * Corresponding author. Tel.:+33-5-5543-5884; fax:+
33-5-5543-5912.
E-mail address:[email protected] (K. Ghanam).
enzyme activities [26]. Some reports on a model of rabbit aortic endothelial cells have shown that estrogen enhanced the activity of Ca2+-activated K+ channels [27]; other studies have demonstrated that acute estro-gen administration in the canine coronary circulation induced epicardial coronary vasodilation through open-ing of adenosine triphosphate (ATP)-sensitive potas-sium channels and calcium channel antagonism [28]. Thus, the relative contribution of EDHF in this benefi-cial effect of estrogen has not been clarified.
Our studies are the first to examine the involvement of EDHF in the protective effect of 17b-estradiol in hypercholesterolemic rabbit carotid arteries, and to de-termine both the nature of EDHF and its target K+ channels, in these arteries, by using selective blockers.
2. Methods
2.1. Arterial ring preparation
Male New Zealand rabbits (2.5 – 3 kg) were assigned randomly to four dietary groups for 8 weeks: control group, normal rabbit diet; (control+estradiol) group, normal rabbit diet+17b-estradiol di-undecylate (intra-muscular (i.m.) injection, 700mg per week); cholesterol group, standard chow+1% cholesterol; and (choles-terol+estradiol) group, 1% cholesterol chow+ 17b-estradiol di-undecylate (i.m. injection, 700mg per week). All animals were sacrificed by exsanguination after anesthesia with sodium pentobarbital (30 mg kg−1 intravenously). The common carotid artery was iso-lated, placed in ice cold Krebs (NaCl, 118.3 mM; KCl, 4.7 mM; CaCl2, 2.5 mM; MgSO4, 1.2 mM; KH2PO4, 1.2 mM; glucose, 11.1 mM; and NaHCO3, 24.9 mM; pH 7.4), cleaned of connective tissue and cut into transverse rings 3 mm long. Special care was taken to avoid damage to the endothelium. Each ring was then suspended vertically in the organ chamber (volume, 25 ml) between two stainless steel hooks, in Krebs solution maintained at 37°C, and gassed with 95% oxygen and 5% carbon dioxide. One of the hooks was fixed to a stand, while the other was attached to an isometric force transducer and the tension recorded on a Gould polygraph model 272. Rings were initially stretched until the resting tension reached 3 g and then allowed to equilibrate for 45 min; during this period, resting tension was continuously monitored and, if necessary, readjusted to 3 g by further stretching.
2.2. Determination of serum cholesterol and triglyceride concentrations
Blood samples were collected in tubes from each animal before excision of the aorta. After centrifuga-tion at 2000×gfor 10 min, the total serum cholesterol
and triglyceride concentrations were measured by a standard enzymatic method (cholesterol enzymatic PAP 61225 for cholesterol and 61236 for triglycerides; Bio Merieux, France).
2.3. Experimental protocols
In a first set of experiments, rings were treated for 40 min with the nitric oxide (NO) synthase inhibitor Nv -nitro-L-arginine methyl ester (L-NAME) (300mM) and the cyclooxygenase inhibitor indomethacin (10 mM). They were then precontracted with phenylephrine (1 mM) and exposed to increasing cumulative concentra-tions of acetylcholine (3 nM – 30 mM). Some carotid artery rings were precontracted with elevated potassium Krebs solution (50 mM), which was made by equimolar replacement of NaCl with KCl. In some experiments, the endothelium was removed by gently rubbing the endothelial surface with a cannula. Removal of the endothelium was confirmed by the absence of a relax-ant response to acetylcholine (3 mM).
To determine the nature of targeted K+ channels, in the presence of L-NAME and indomethacin, either tetraethylammonium (10 mM), charybdotoxin (50 nM) alone or with apamin (50 nM), glibenclamide (10 mM) or 4-aminopyridine (1 mM) were added 40 min before testing the acetylcholine (ACh) effect, to block, respec-tively, calcium-activated K+, ATP-sensitive K+ and voltage-dependent delayed rectifier K+ channels.
In another set of experiments, in the presence of L-NAME and indomethacin, the rings were preincu-bated with hemoglobin (10 mM), a nitric oxide scav-enger, LY83183 (10 mM), a specific inhibitor of guanylate cyclase or clotrimazole (1 mM) and an in-hibitor of cytochrome P450-monooxygenase.
In all experiments, the phenylephrine concentration was adjusted in order to obtain a similar amount of precontraction.
2.4. Statistical analysis
2.5. Drugs
Phenylephrine, acetylcholine, Nv
nitro-L-arginine methyl ester, indomethacin, tetraethylammonium, charybdotoxin, apamin, 4-aminopyridine, hemoglobin, LY83183 and clotrimazole were purchased from Sigma Chemical Co. Glibenclamide was purchased from Re-search Biochemicals Inc. 17b-Estradiol di-undecylate was purchased from Theramex. Stock solutions of phenylephrine, acetylcholine,L-NAME, charybdotoxin, apamin and 4-aminopyridine were diluted in distilled water. Clotrimazole, LY83183 and indomethacin were dissolved in ethanol. Glibenclamide was diluted in dimethylsulphoxide. The final concentrations of all sol-vents used (B0.1%) had no effects in preliminary ex-periments. 17b-Estradiol di-undecylate was emulsioned in vegetable oil.
3. Results
3.1. Serum cholesterol and triglyceride le6els
Serum cholesterol and triglyceride levels did not dif-fer between control groups treated or not with 17b-estradiol. Serum total cholesterol levels were markedly elevated in groups fed a high cholesterol diet, whether or not treated with 17b-estradiol. In the (cholesterol+
estradiol) group, the serum total cholesterol level was significantly lower than in the cholesterol group. Serum triglyceride levels were also significantly greater in the cholesterol group compared with the control group, but remained practically unchanged in the (cholesterol+
estradiol) group (Table 1).
3.2. Effects of acetylcholine
Acetylcholine (3 nM – 30 mM) caused a concentra-tion-dependent relaxation in carotid artery precon-tracted with 1 mM phenylephrine. Neither hypercholesterolemia nor treatment with 17b-estradiol affected significantly this relaxation (Fig. 1).
Fig. 1. Effect of acetylcholine in the rabbit carotid artery, precon-tracted with phenylephrine (1mM), in the absence or in the presence ofL-NAME (300mM), and indomethacin (10mM) in control, (con-trol+estradiol), cholesterol and (cholesterol+estradiol) groups. Points are the mean from eight separate experiments, vertical lines show standard error of the mean.
3.3. L-NAME and indomethacin-insensiti6e relaxation
induced by acetylcholine
In all groups, removal of the endothelium or precon-traction of the arteries by elevated external K+ (50 mM) completely abolished the L-NAME and in-domethacin-insensitive relaxation induced by acetyl-choline. Neither L-NAME (300mM) nor indomethacin (10 mM) modified significantly the level of phenylephrine-induced precontraction.
3.3.1. Control group
In the presence of L-NAME and indomethacin, acetylcholine (3 nM – 30 mM) induced a concentration-dependent relaxation of endothelium-intact rings from rabbit carotid arteries. The pD2 value was 6.3490.05 and the maximal response (Em) reached 30.893.5% (n=8) (Fig. 1).
3.3.2. Control group treated with 17b-estradiol
In the presence ofL-NAME and indomethacin, treat-ment with 17b-estradiol did not modify significantly the acetylcholine response of carotid arteries isolated from the control group. The pD2 value was 6.7490.04 (ver-sus 6.3490.05), and the maximal response (Em) was 33.492.9% (versus 30.893.5%) (n=8) (Fig. 1).
3.3.3. Cholesterol group
The concentration – response curve to ACh was sig-nificantly shifted to the right compared with the control Table 1
Cholesterol and triglyceride levelsa
Total cholesterol Triglycerides (mM) Group
(mM)
2.090.7 Control 1.690.3
1.890.2
(Control+estradiol) 1.990,4 47.993.3* 11.392.0* Cholesterol
2.690.5c
34.993.2*,c
(Cholesterol
+estradiol)
aEach value is the mean9S.E.M. from eight animals.
* Significantly different versus the control group (PB0.05).
c
group. Em value was decreased to 12.291.7%, while the pD2 value was not significantly modified: 6.369 0.14 (versus 6.3490.05) (n=8) (Fig. 1).
3.3.4. Cholesterol group treated with 17b-estradiol The ACh-induced vasorelaxation was similar to the control group: the pD2 value was 6.5390.06 (versus 6.3490.05) (n=8) and theEmwas 29.593.9% (versus 30.893.5%) (n=8) (Fig. 1).
3.4. Effects of tetraethylammonium, glibenclamide, 4 -aminopyridine, apamin+charybdotoxin on L-NAME and indomethacin-insensiti6e relaxation to acetylcholine
3.4.1. Control group
Tetraethylammonium inhibited partially, but signifi-cantly, the L-NAME/indomethacin-insensitive relax-ation to ACh. The pD2 value was 5.9490.05 (versus 6.3490.05) (n=8) and the maximal response was 16.894.8% (versus 30.893.5%) (n=8). Neither glibenclamide nor 4-aminopyridine affected signifi-cantly this residual relaxation to ACh. The pD2 values were, respectively, 6.6190.04 and 6.3090.14 (versus 6.3490.05) (n=8), while the maximal responses were, respectively, 25.995.5 and 34.296.7% (versus 30.89
3.5%) (n=8). Charybdotoxin, a blocker of large-con-ductance Ca2+
-activated potassium channels (BKCa), completely inhibited this response. Addition of apamin, a blocker of small-conductance Ca2+-activated potas-sium channels (SKCa), to charybdotoxin did not signifi-cantly modify the response to ACh (Fig. 2).
3.4.2. Control group treated with 17b-estradiol
Like the control group, in the presence of L-NAME and indomethacin, 4-aminopyridine did not modify re-laxation to ACh; the pD2value was 6.4690.02 (versus 6.3490.05) (n=8) and the maximal response was 32.193.4% (versus 30.893.5%) (n=8). Tetraethylam-monium reduced the L-NAME/indomethacin-resistant relaxation to ACh; the pD2 value was 6.6190.04 (ver-sus 6.3490.05) (n=8) and the maximal response was 22.192.2% (versus 30.893.5%) (n=8). Charybdo-toxin, alone or in the presence of apamin, abolished this relaxation. However, unlike the control group, glibenclamide abolished this relaxation in the control group treated with 17b-estradiol; the pD2 value was 5.9490.05 (versus 6.3490.05) (n=8) and the maximal response was 6.290.5% (versus 30.893.5%) (n=8) (Fig. 3).
3.4.3. Cholesterol group
Neither glibenclamide nor 4-aminopyridine modified significantly theL-NAME/indomethacin-resistant relax-ation to ACh. The pD2 values were, respectively, 6.4690.10 and 6.6790.07 (versus 6.3690.14) (n=8). Similarly, the maximal responses were not significantly
different:Em, 12.690.4 and 11.591.4% (versus 12.29 1.7%) (n=8). However, tetraethylammonium reduced partially the L-NAME/indomethacin-resistant relax-ation. The pD2 value was 5.9090.14 (versus 6.369 0.14) (n=8) and the maximal response was 8.190.1% (versus 12.291.7%) (n=8). Charybdotoxin alone or with apamin completely abolished this relaxation (Fig. 4).
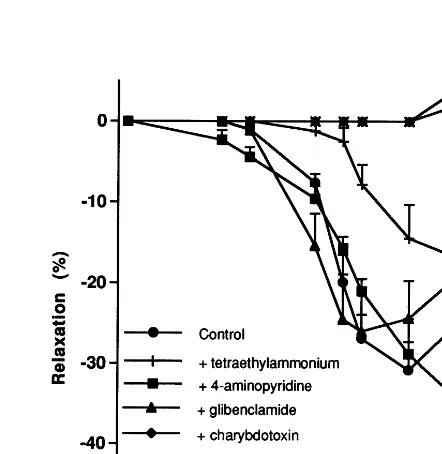
3.4.4. Cholesterol group treated with 17b-estradiol Pretreatment with 4-aminopyridine did not induce a statistically significant change of the L-NAME/ in-domethacin-insensitive relaxation to ACh, pD2= 6.1490.09% (versus 6.5390.06%) (n=8). In contrast, both tetraethylammonium, glibenclamide and charyb-dotoxin alone or with apamin practically abolished this relaxation (Fig. 5).
3.5. Effect of hemoglobin, LY83183 or clotrimazole on
L-NAME/indomethacin-insensiti6e relaxation to acetyl
-choline
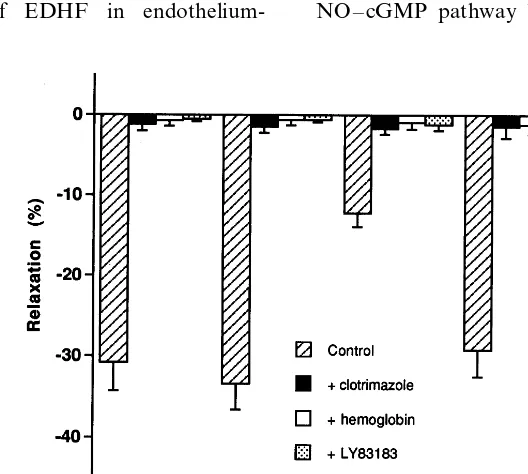
In all groups, in the presence of L-NAME+ in-domethacin, pretreatment with hemoglobin (10 mM), LY83183 (10 mM) or clotrimazole (1 mM), completely abolished the ACh-induced relaxation (Fig. 6).
Fig. 2. Mean concentration – response curves for acetylcholine in the rabbit carotid artery, precontracted with phenylephrine (1mM) and isolated from the control group in the presence of tetraethylammo-nium (1 mM), charybdotoxin (50 nM), alone or with apamin (50 nM), 4-aminopyridine (1 mM) or glibenclamide (10 mM). Arteries were preincubated with L-NAME (300 mM) and indomethacin (10
Fig. 3. Mean concentration – response curves for acetylcholine in the rabbit carotid artery, precontracted with phenylephrine (1mM) and isolated from the (control+estradiol) group, in the presence of tetraethylammonium (1 mM), charybdotoxin (50 nM), alone or with apamin (50 nM), 4-aminopyridine (1 mM) or glibenclamide (10mM). Arteries were preincubated with L-NAME (300 mM) and in-domethacin (10mM). Points are the mean from eight separate exper-iments, vertical lines show standard error of the mean.
nitroprusside) were not altered by high-cholesterol diet for 8 weeks. Contractile responses to phenylephrine (receptor-dependent vasoconstrictor) or high K+ solu-tion (voltage-dependent vasoconstrictor) were also not significantly modified in 8-week dietary cholesterol rab-bit arteries compared with control arteries. These re-sults suggest that the severity of effects of hypercholesterolemia depended on the type of the artery.
Furthermore, several data have shown a protective effect of chronic treatment with 17b-estradiol in hyperc-holesterolemic blood vessels. This effect was at-tributable to its favorable effects on lipid profile [25] or its direct vascular effects by a regulation of the produc-tion and the release of NO [18] or prostacyclin [29]. In contrast, little is known about the effects of estrogen on the production of EDHF. In our data, treatment by 17b-estradiol for 8 weeks attenuated the serum choles-terol level in hypercholescholes-terolemic rabbits (Table 1), confirming the beneficial effect of estrogen on the plasma lipids. The new finding of this study is the observation of an additional mechanism for the protec-tive effect of chronic treatment by 17b-estradiol against hypercholesterolemia. In our experiments, we observed a L-NAME/indomethacin-resistant relaxation to acetyl-choline in carotid arteries isolated from control rabbits. This relaxation was suppressed by removal of the en-dothelium, by elevated external K+ or by addition of charybdotoxin, suggesting the presence of an EDHF-4. Discussion.
In our data, 8-week feeding of rabbits with 1% cholesterol induced an increase of the plasma choles-terol concentrations. This hypercholescholes-terolemia was as-sociated with a liver injury: the livers were pale yellow, had a finely nodular surface and were characterized by a severe fibrosis. It has been clearly established that the hypercholesterolemic diet caused marked alterations in the plasma lipoproteins, particularly an increase of low-density lipoprotein (LDL) and a decrease of high-density lipoprotein. The liver being the main organ of LDL catabolism and having receptors for the persistent excess LDL, this liver injury developed concomitant with arterial injury. In our preliminary studies (data not shown), we observed atheromatous plaques and modifi-cations of vascular reactivity of many rabbit arteries. These plaques were more important in the thoracic aorta than in the abdominal aorta, but were absent in the carotid, middle cerebral and saphenous arteries. Additionally, these arteries presented modifications of responsiveness to vasodilators but not to vasoconstric-tors. Indeed, hypercholesterolemia did not modify the response to acetylcholine in the carotid artery, but reduced it by 28% in the saphenous artery, by 30% in the middle cerebral artery, by 33% in the abdominal aorta (not shown) and by 50% in the thoracic aorta [26]. In contrast, vasodilatory responses of all these arteries to an endothelium-independent agonist (sodium
Fig. 4. Mean concentration – response curves for acetylcholine in the rabbit carotid artery, precontracted with phenylephrine (1mM) and isolated from the cholesterol group in the presence of tetraethylam-monium (1 mM), charybdotoxin (50 nM), alone or with apamin (50 nM), 4-aminopyridine (1 mM) or glibenclamide (10 mM). Arteries were preincubated with L-NAME (300 mM) and indomethacin (10
Fig. 5. Mean concentration – response curves for acetylcholine in the rabbit carotid artery, precontracted with phenylephrine (1mM) and isolated from the (cholesterol+estradiol) group, in the presence of tetraethylammonium (1 mM), charybdotoxin (50 nM), alone or with apamin (50 nM), 4-aminopyridine (1 mM) or glibenclamide (10mM). Arteries were preincubated with L-NAME (300 mM) and in-domethacin (10mM). Points are the mean from eight separate exper-iments, vertical lines show standard error of the mean.
dependent relaxation induced by acetylcholine on the same artery. Furthermore, in our study, on rabbits fed high cholesterol, this L-NAME/ indomethacin-insensi-tive relaxation to ACh was significantly reduced, sug-gesting that this response may be impaired by hypercholesterolemia. The work of Urakami-Harasawa et al. [23] supports this hypothesis, and shows that aging and hypercholesterolemia significantly reduced EDHF-mediated relaxation evoked by ACh and bradykinin in large human arteries. Similarly, Cowan and Steffen [22] showed that treatment of the rabbit abdominal aorta with lysophosphatidylcholine, which plays an important role in promoting the atherogenic process, inhibited the relaxation mediated by EDHF. Furthermore, in the rabbit carotid artery, Najibi et al. [31] demonstrated that hypercholesterolemia for 10 weeks did not affect the relaxations to ACh and proved that this normal relaxation occured, despite the absence of NO-mediated increased cGMP, by activation of charybdotoxin-sensitive potassium channels. Our exper-iments confirmed these data, since no significant modifi-cation of the relaxation responses to ACh was observed in the cholesterol group, compared with the control group (Fig. 1). Nevertheless, in the presence of L -NAME and indomethacin, the residual relaxation to ACh was reduced in hyperchosterolemic arteries. This response was abolished by a NO scavenger such as hemoglobin or an inhibitor of soluble guanylate cyclase such as LY83183 (Fig. 5), suggesting a reduction of the NO – cGMP pathway by hypercholesterolemia. In the dependent mechanism in the ACh response. These
re-sults are in close agreement with recent data [30] indi-cating a contribution of EDHF in
endothelium-Fig. 6. Effect of hemoglobin (10mM), LY83183 (10mM) or clotrimazole (1mM) on the maximal response to acetylcholine, in the rabbit carotid artery, isolated from control, (control+estradiol), cholesterol and (cholesterol+estradiol) groups. The arteries were treated withL-NAME (300
presence ofL-NAME and indomethacin, the treatment by 17b-estradiol during 8 weeks allowed relaxation to ACh, which was, otherwise impaired by the high-cholesterol diet. This vasorelaxation was abolished in the presence of potassium chloride. This is a novel finding that supports the hypothesis that chronic estro-gen treatment protects hypercholesterolemic rabbit carotid arteries by an EDHF-mediated mechanism. The estrogen treatment probably increases the release of EDHF from the endothelium of hypercholesterolemic rabbit carotid arteries.
The chemical identity of EDHF remains elusive. In our study, in all groups, hemoglobin, abolished this EDHF-mediated response, suggesting that nitric oxide mediated this remaining relaxation. Previous studies [14,15] reported that NO induced hyperpolarization. In the rabbit isolated carotid artery, Cohen et al. [16] have demonstrated that high concentrations ofL-NAME (30 mM) did not fully block endothelium-relaxation and hyperpolarization, and proved that NO, rather than a different factor, was the mediator of ACh-induced en-dothelium-dependent relaxation and hyperpolarization in this artery. In recent data, in rat superior mesenteric artery, Simonsen et al. [32] confirmed these results, and showed that NO mediated theL-NORG-resistant relax-ations to ACh. Thus, in our experiments, it is likely that NO mediated the indomethacin/L -NAME-insensi-tive relaxation to ACh. In addition, LY83183, a specific inhibitor of guanylate cyclase, completely blocked this residual relaxation to ACh in the presence of NO synthase and cyclooxygenase inhibitors, suggesting that activation of guanylate cyclase may be one mechanism by which NO evoked this residual relaxation to ACh. Therefore, the protective effect of estrogen on hyperc-holesterolemic carotid artery is mediated by a NO – cGMP pathway. These results agree with several studies [24,33,34], in which estradiol can increase the produc-tion of endothelium-derived nitric oxide. However, in all groups, clotrimazole, an inhibitor of cytochrome P450-monooxygenase, abolished the L-NAME/ in-domethacin-insensitive relaxing response to ACh in the carotid artery. This result could suggest that this relax-ation involves a cytochrome P450-derived arachidonic acid metabolite. Some studies [29,35] have proposed that EDHF may be a cytochrome P450 product in rabbit carotid artery. Nevertheless, the site of action of clotrimazole is controversial. Rittenhouse et al. [36] have shown that clotrimazole is a nonspecific cy-tochrome P450 inhibitor and could be a potent direct inhibitor of calcium-dependent potassium channels. Other authors proved that this compound inhibits not only EDHF-mediated relaxations, but also response to potassium channel openers [37] and endothelium-derived NO [38]. Thus, in our study, clotrimazole prob-ably inhibited the activation of potassium channels by nitric oxide.
Furthermore, to investigate the implication of K+ channels in the protective effect of 17b-estradiol on hypercholesterolemic carotid artery, different K+ chan-nels blockers were used. In the carotid artery isolated from control and hypercholesterolemic rabbits, charyb-dotoxin alone or with apamin, abolished theL-NAME/ indomethacin-resistant relaxation to acetylcholine, while neither 4-aminopyridine nor glibenclamide af-fected this response. This suggests that only Ca2+ -acti-vated K+channels are involved in the EDHF-mediated relaxation in these groups. These results agree with recent work on the carotid artery isolated from control rabbits [29] and from hypercholesterolemic rabbits [30], which showed that charybdotoxin completely inhibited the EDHF-mediated relaxations to ACh. In control and hypercholesterolemic rabbits treated with 17b-estradiol, charybdotoxin alone or with apamin or glibenclamide suppressed this indomethacin- and L -NAME-resistant endothelium-dependent relaxation to ACh, suggesting that at least two kinds of K+channels are involved in the effect of 17b-estradiol: Ca2+ -depen-dent and ATP-sensitive potassium channels. Our results are reminiscent of those by Rusko et al. [27], which showed that the acute effect of 17b-estradiol was to markedly enhance the activity of the Ca2+-activated K+channels on endothelial cells of rabbit aorta. White et al. [39] also found that 17b-estradiol relaxes porcine coronary arteries by opening KCa channels via cGMP-dependent phosphorylation. In accordance, Node et al. [40] have also demonstrated that 17b-estradiol reduces myocardial infarct size and the incidence of ischemia in the canine heart, by a NO pathway and the opening of KCachannels. There is, thus, strong evidence to support the activation of KCa channels by estrogen. Neverthe-less, an interesting finding in the present study is that the specific KATP antagonist, glibenclamide, also abol-ished the relaxation to ACh in the control and choles-terol groups treated with estrogen, suggesting that ATP-sensitive potassium channels may be other candi-dates involved in the estrogenic action on carotid arter-ies. In previous work, Sudhir et al. [28] showed that acute estrogen administration on canine epicardial coronary arteries induced a vasodilatory response. This effect was reduced by pretreatment with glibenclamide, suggesting that opening of ATP-sensitive potassium channels may be a mechanism of this vasodilator effect. Additionally, our data indicates that chronic treatment by 17b-estradiol on hypercholesterolemic rabbit carotid arteries activates both the ATP-sensitive and Ca2+ -ac-tivated potassium channels, since either glibenclamide or charybdotoxin abolish the restored relaxation to ACh.
effect appears to be mediated by release of nitric oxide, which activates calcium- and ATP-dependent potas-sium channels via a cyclic GMP pathway. Further studies are required to show whether EDHF is involved in this beneficial effect of 17b-estradiol in other arteries.
Acknowledgements
This study was supported by a grant from the Limousin region.
References
[1] Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res 1987;61:866 – 79.
[2] Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relax-ing factor. Nature 1987;327:524 – 6.
[3] Moncada S, Vane JR. Pharmacology and endogenous roles of prostaglandins, endoperoxides, thromboxane A2 and prostacy-clin. Pharmacol Rev 1978;30:293 – 331.
[4] Chen GF, Suzuki H. Calcium dependency of the endothelium-dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. J Physiol 1990;421:521 – 34.
[5] Nagao T, Vanhoutte PM. Endothelium-derived hyperpolarizing factor and endothelium-dependent relaxations. Am J Respir Cell Mol Biol 1993;8:1 – 6.
[6] Mombouli JV, Vanhoutte PM. Endothelium-derived hyperpolar-izing factor (s): updating the unknown. Trends Pharmacol Sci 1997;18:252 – 6.
[7] Corriu C, Feletou M, Canet E, Vanhoutte PM. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br J Pharmacol 1996;119:959 – 64.
[8] Petersson J, Zygmunt PM, Hogestatt ED. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br J Pharmacol 1997;120:1344 – 50. [9] Cowan CL, Palacino JJ, Najibi S, Cohen RA. Potassium
chan-nel-mediated relaxation to acetylcholine in rabbit arteries. J Pharmacol Exp Ther 1993;266:1482 – 9.
[10] Garland CJ, Plane F, Kemp BK, Cocks TM. Endothelium-de-pendent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol Sci 1995;16:23 – 30.
[11] Campbell WB, Gebremedhin D, Pratt PH, Harder DR. Identifi-cation of epoxyeicosatrienoic acids as endothelium-derived hy-perpolarizing factors. Circ Res 1996;78:415 – 23.
[12] Randall MD, Alexander SP, Bennet T, Boyd EA, Fry JR, Gardiner SM, Kemp PA, McCulloch AI, Kendall DA. An endogenous cannabinoid as an endothelium-derived vasorelax-ant. Biochem Biophys Res Commun 1996;229:114 – 20. [13] Edwards C, Dora KA, Gardener MJ, Garland CJ, Weston AH.
K+ is an endothelium-derived hyperpolarizing factor in rat
arteries. Nature 1998;396:269 – 72.
[14] Tare M, Parkington HC, Coleman HA, Neild TO, Dusting GJ. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from endothelium. Nature 1990;346:69 – 71.
[15] Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994;368:850 – 3.
[16] Cohen RA, Plane F, Najibi S, Huk I, Malinski T, Garland CJ. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc Natl Acad Sci USA 1997;94:4193 – 8.
[17] Jayakody L, Senaratne M, Thomson A, Kappagoda T. Endothe-lium-dependent relaxation in experimental atherosclerosis in the rabbit. Circ Res 1987;60:251 – 64.
[18] Williams JK, Adams MR, Herrington DM, Clarkson TB. Short term administration of estrogen and vascular responses of atherosclerotic coronary arteries. J Am Coll Cardiol 1992;20:452 – 7.
[19] Harrison DG, Ohara Y. Physiological consequences of increased vascular oxidant stresses in hypercholesterolemia and atherscle-rosis: implications for impaired vasomotion. Am J Cardiol 1995;75:75B – 81B.
[20] Minor RL, Myers PR, Guerra R, Bates JN, Harrison DG. Diet-induced atherosclerosis increases the release of nitrogen oxides from rabbit aorta. J Clin Invest 1990;86:2109 – 16. [21] White CR, Brock TA, Chang LY, Crapo J, Briscoe P, Ku D,
Bradley WA, Gianturco SH, Gore J, Freeman BA. Superoxide and peroxynitrite in atherosclerosis. Proc Natl Acad Sci USA 1994;91:1044 – 8.
[22] Cowan CL, Steffen RP. Lysophosphatidylcholine inhibits relax-ation of rabbit abdominal aorta mediated by endothelium-derived nitric oxide and endothelium-endothelium-derived hyperpolarizing factor independent of protein kinase C activation. Arterioscler Thromb Vasc Biol 1995;15:2290 – 7.
[23] Urakami-Harasawa L, Shimokawa H, Nakashima M, Egashira K, Takeshita A. Importance of endothelium-derived hyperpolar-izing factor in human arteries. J Clin Invest 1997;100:2793 – 9. [24] Hayashi T, Yamada K, Esaki T, Mutoh E, Iguchi A. Effect of
estrogen on isoforms of nitric oxide synthase: possible mecha-nism of anti-atherosclerotic effect of estrogen. Gerontology 1997;43:24 – 34.
[25] Keaney JF, Shwaery GT, Xu A, Nicolosi RJ, Loscalzo J, Foxall TL, Vita JA. 17b-Estradiol preserves endothelial vasodilator function and limits low-density lipoprotein oxidation in hyperc-holesterolemic swine. Circulation 1994;89:2251 – 9.
[26] Ghanam K, Javellaud J, Ea-Kim L, Oudart N. The protective effect of 17b-estradiol on vasomotor responses of aorta from cholesterol-fed rabbit is reduced by inhibitors of superoxide dismutase and catalase. Biochem Biophys Res Commun 1998;249:858 – 64.
[27] Rusko J, Li L, Van Breemen C. 17-b-Estradiol stimulation of endothelial K+ channels. Biochem Biophys Res Commun
1995;214:367 – 72.
[28] Sudhir K, Chou TM, Mullen WL, Hausmann D, Collins P, Yock PG, Chatterjee K. Mechanisms of estrogen-induced va-sodilation: in vivo studies in canine coronary conductance and resistance arteries. J Am Coll Cardiol 1995;26:807 – 14. [29] Fogelberg M, Vesterqvist O, Diczfalusy U, Henriksson P.
Exper-imental atherosclerosis: Effects of oestrogen and atherosclerosis on thromboxane and prostacyclin formation. Eur J Clin Invest 1990;20:105 – 10.
[30] Dong H, Waldron GJ, Galipeau D, Cole WC, Triggle CR. NO/PGI2-independent vasorelaxation and the cytochrome P450 pathway in rabbit carotid artery. Br J Pharmacol 1997;120:695 – 701.
[31] Najibi S, Cowan CL, Palacino JJ, Cohen RA. Enhanced role of potassium channels in relaxations to acetylcholine in hyperc-holesterolemic rabbit carotid artery. Am J Physiol 1994;266:H2061 – 7.
[33] Andersen HL, Weis JU, Fjalland B, Korsgaard N. Effect of acute and long-term treatment with 17b-estradiol on the vasomo-tor responses in the rat aorta. Br J Pharmacol 1999;126:159 – 68. [34] Nekooeian AA, Lim SL, Man RYK, Pang CCY. Chronic 17b -estradiol augments relaxant role of basal nitric oxide in blood vessels from rats with heart failure. Naunyn-Schmiedeberg’s Arch Pharmacol 1999;358:671 – 7.
[35] Lischke V, Busse R, Hecker M. Selective inhibition by barbitu-rates of the synthesis of endothelium-derived hyperpolarizing factor in the rabbit carotid artery. Br J Pharmacol 1995;115:969 – 74.
[36] Rittenhouse AR, Vandorpe DH, Brugnara C, Alper SL. The antifungal imidazole clotrimazole is potent blocker of calcium-activated potassium channels. J Membr Biol 1997;157:177 – 92.
[37] Vanheel B, Van de Voorde J. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hy-perpolarization of the rat main mesenteric artery. J Physiol (Lond) 1997;501:331 – 41.
[38] Asbun J, Escalante B, Ceballos G, Ocharan ME, Castillo EF, Castillo C. Clotrimazole inhibits nitric oxide-mediated-relaxation elicited by acetylcholine in rat aortic rings. Proc West Pharmacol Soc 1998;41:145 – 6.
[39] White RE, Darkow DJ, Lang JL. Estrogen relaxes coronary arteries by opening BKCa channels through a cGMP-dependent mechanism. Circ Res 1995;77:936 – 42.
[40] Node K, Kitakaze M, Kosaka H, Minamino T, Funaya H, Hori M. Amelioration of ischemia- and reperfusion-induced myocar-dial injury by 17b-estradiol: role of nitric oxide and calcium-acti-vated potassium channels. Circulation 1997;96:1953 – 63.