The effects of lifibrol (K12.148) on the cholesterol metabolism of
cultured cells: evidence for sterol independent stimulation of the
LDL receptor pathway
Hubert Scharnagl
a,*, Michael Schliack
b, Roland Lo¨ser
b, Markus Nauck
a,
Hedi Gierens
a, Nikola Jeck
c, Heinrich Wieland
a, Werner Groß
c, Winfried Ma¨rz
aaDepartment of Medicine,Di6ison of Clinical Chemistry,Albert Ludwigs-Uni6ersity,Hugstetter Strasse55,79106Freiburg,Germany bDepartment of Biochemistry,Klinge Pharma GmbH,Munich,Germany
cGusta6Embden-Center of Biological Chemistry,Johann Wolfgang Goethe-Uni6ersity,Frankfurt,Germany
Received 23 September 1999; accepted 25 January 2000
Abstract
Lifibrol (4-(4%-tert. butylphenyl)-1-(4%-carboxyphenoxy)-2-butanol) is a new hypocholesterolemic compound; it effectively lowers low density lipoprotein (LDL) cholesterol. We studied the effects of lifibrol on the cholesterol metabolism of cultured cells. In the hepatoma cell line HepG2, Lifibrol decreased the formation of sterols from [14C]-acetic acid by approximately 25%. Similar to lovastatin, lifibrol had no effect on the synthesis of sterols from [14C]-mevalonic acid. Lifibrol did not inhibit 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase. Instead, cholesterol synthesis inhibition by lifibrol was entirely accounted for by competitive inhibition of HMG-CoA synthase. Lifibrol enhanced the cellular binding, uptake, and degradation of LDL in cultured cells in a dose dependent fashion. The stimulation of LDL receptors was significantly stronger than expected from the effect of lifibrol on sterol synthesis. In parallel, lifibrol increased the amount of immunologically detectable receptor protein. Stimulation of LDL receptor mediated endocytosis was observed both in the presence and in the absence of cholesterol-containing lipoproteins. In the absence of an extracellular source of cholesterol, both lifibrol and lovastatin induced microsomal HMG-CoA reductase. Co-incubation with LDL was sufficient to suppress the lifibrol mediated increase in reductase activity, indicating that lifibrol does not affect the production of the non-sterol derivative(s) which are thought to regulate HMG-CoA reductase activity at the post-transcriptional level. Considered together, the data suggest that the hypolipidemic action of lifibrol may, at least in part, be mediated by sterol-independent stimulation of the LDL receptor pathway. A potential advantage of lifibrol is that therapeutic concentrations do not interfere with the production of mevalonate which is required not only to synthesize sterols but also as a precursor of electron transport moieties, glycoproteins and farnesylated proteins. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Hypercholesterolemia; Cholesterol biosynthesis; 3-Hydroxy-3-methylglutaryl coenzyme A synthase; 3-Hydroxy-3-methylglutaryl coen-zyme A reductase; Low density lipoprotein receptor; Lifibrol (K12.148); Atherosclerosis
www.elsevier.com/locate/atherosclerosis
1. Introduction
There is now abundant evidence that hypercholes-terolemia due to high concentrations of apolipoprotein
B containing lipoproteins is one of the major risk factors of atherosclerosis and that the reduction of LDL cholesterol lowers the cardiovascular risk [1 – 5].
Abbre6iations: Ac-CoA, acetyl-CoA; AcAc-CoA, acetoacetyl-CoA; cpm, counts per minute; DMSO, dimethyl sulfoxide; EDTA, ethylenedi-aminetetraacetate; FBS, fetal bovine serum; HMG, 3-hydroxy-3-methylglutaric acid; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A;Km,
Michaelisconstant; Lifibrol, 4-(4%-tert. butylphenyl)-1-(4%-carboxyphenoxy)-2-butanol; LDL, low density lipoproteins; LPDS, human lipoprotein deficient serum; PBS, phosphate buffered saline; PMSF, phenylmethylsulfonylfluoride; SDS, sodium dodecyl sulfate; Tris, tris(hydroxy-methyl)aminomethane;Vmax, maximum reaction rate.
* Corresponding author. Tel.: +49-761-2707306; fax: +49-761-2703444. E-mail address:[email protected] (H. Scharnagl).
The steady-state concentration of low density lipo-proteins (LDL) is mainly regulated by LDL receptors on the surface of liver cells, which are responsible for approximately 75% of the catabolism of LDL in the body [6]. In patients lacking functional LDL receptors, not only is the fractional catabolic rate of LDL de-creased, but also the production of LDL is increased. This is due to the fact that LDL receptors normally bind and internalize remnants of triglyceride-rich lipo-proteins which serve as precursors for the formation of LDL [7]. The expression of LDL receptors is finely tuned according to the cell’s demand for cholesterol. In the absence of extracellular sources of cholesterol, cells enhance the production of LDL receptors, together with coordinate increases in HMG-CoA reductase and HMG-CoA synthase, the two rate-limiting enzymes of the sterol biosynthesis pathway [6,8]. Similar responses are elicited in cells in which the endogenous production of sterols is inhibited, for instance by competitive HMG-CoA reductase inhibitors.
The most successful strategies to reduce the concen-tration of LDL in the circulation have in common that they involve the up-regulation of the LDL receptor activity by depleting the regulatory pool of cholesterol in the liver. Anion exchanging resins reduce the reab-sorption of bile salts from the intestine and stimulate the conversion of cholesterol to bile acids, plant sterols interfere with the absorption of cholesterol from the intestine, and the HMG-CoA reductase inhibitors re-duce the de no6o production of sterols [9,10].
Lifibrol is a novel, highly effective lipid-lowering agent. The hypolipidemic properties of Lifibrol have been demonstrated in rats, marmosets, WHHL-rabbits and pigs [11,12]. Lifibrol strongly reduced serum choles-terol and triglycerides in these studies. In hyperlipi-demic humans, lifibrol lowered LDL cholesterol and triglycerides by approximately 40 and 25%, respectively [13 – 15]. In all trials reported so far, the maximum hypocholesterolemic effect occurred earlier than with 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, nicotinic acid or bile acid seques-trants [16 – 18] and was sustained during the entire administration period. The rapid lipid lowering effect of lifibrol strongly suggests that its mechanism of action is distinct from that of the HMG-CoA reductase in-hibitors. Another interesting feature of lifibrol is that it brought about substantial decreases of Lp(a) and fibrinogen [15]. There has been evidence that lifibrol acts by inhibiting cholesterol biosynthesis [19]. Recently published studies examined the effect of lifibrol on the in vivo metabolism of apolipoprotein B. These data suggest that lifibrol lowers cholesterol by increasing the receptor-mediated catabolism of LDL [20].
The purpose of this study was to examine in detail the effects of lifibrol on the cholesterol metabolism of cultured cells. The results provide evidence that lifibrol
enhances the expression of membrane LDL receptors independent from the cellular regulatory cholesterol pool and suggest that lifibrol sets up a new class of hypolipidemics with a mode of action clearly distinct from the fibrates and from competitive inhibitors of HMG-CoA reductase.
2. Materials and methods
2.1. Materials
Lifibrol (4-(4%-tert. butylphenyl)-1-(4% -carboxyphe-noxy)-2-butanol) was synthesized at Klinge Pharma GmbH (Munich, Germany). Lovastatin was from Merck Sharp and Dohme. The lactone form of lova-statin was converted into the sodium salt according to the procedure of Cutts et al. [21]. Stock solutions containing 10 mM of lifibrol (in DMSO) or lovastatin (in ethanol/NaOH) were stored at −20°C until use. The final concentration of DMSO did not exceed 0.1% (vol/vol). [2-14C]-acetic acid, sodium salt (56 mCi/ mmol), [1-14C]-Ac-CoA (specific activity 50 – 62 mCi/ mmol), [1,2-3H]-cholesterol (52 Ci/mmol), DL-3 -hydroxy-3-methyl-[3-14
C]-glutaryl coenzyme A (58 mCi/mmol), DL-[2-3
H]-mevalonic acid lactone (1.26 mCi/mmol), [2-14
C]-mevalonic acid lactone (58 mCi/
mmol) and Na[125I] (carrier free, 17 Ci
/mg) were from Du Pont New England Nuclear (Bad Homburg, Ger-many). Silica gel thin layer chromatography plates (20×20 cm) on aluminium sheet support were from E. Merck (Darmstadt, Germany). ITLC-SA polysilic glass fiber sheets (20×20 cm) were from Gelman Sciences (Ann Arbor, MI). Scintillation fluid (Quicksafe A) was from Zinsser Analytik (Frankfurt, Germany). Polystyrene tissue culture flasks and CovaLink multi-well plates were from Nunc (Roskilde, Denmark). Cell culture medium, additives (glutamine, penicillin G, streptomycin) and fetal bovine serum (FBS) were from Gibco/BRL, (Eggenstein, Germany). Glucose-6-phos-phate dehydrogenase (EC 1.1.1.49, 350 U/mg) was from Boehringer Mannheim (Mannheim, Germany). The LDL receptor specific monoclonal antibody C7 was from Amersham (Braunschweig, Germany). Biotiny-lated goat anti-mouse antibody and avidin:biotinyBiotiny-lated horseradish peroxidase reagent (ABC Peroxidase Elite) were provided by Vector Laboratories (Burlingame, CA).
2.2. Lipoproteins and serum
lipoprotein-deficient serum (LPDS) was prepared by ultracentrifugation as described [24] and stored at −
25°C.
2.3. Cell culture
HepG2 were obtained from the American Type Cul-ture Collection (Rockville, MD). Human skin fibro-blasts were from skin biopsies of normolipidemic individuals. The cells were grown in 80 cm2 flasks containing RPMI 1640 medium supplemented with penicillin G (100 000 units/l), streptomycin (100 mg/l) and 10% (vol/vol) FBS in a humidified incubator at 5% (vol/vol) CO2 and 37°C.
2.4. Incorporation of acetate or me6alonate into
cellular sterols
HepG2 cells were seeded in 25 cm2 flasks, each well containing 5 ml medium with FBS and grown to 70% confluence. Forty hours before the experiments, the cells were washed with PBS and switched to medium supplemented with 10% (vol/vol) human LPDS. The monolayers were then incubated for 6 h with lifibrol or lovastatin at the indicated concentrations. Two hours before the end of the incubation period, the cells were pulse-labelled with [14
C]-acetate or [14
C]-mevalonate both at final concentrations of 35 mM (2 mCi/l medium). After incubation, the cells were washed three times with 3 ml of 150 mM NaCl and suspended in 3 ml n-hexane:isopropanol (3:2 by volume). After adding 0.25 mCi of [1,23-H]-cholesterol, each monolayer was extracted for 30 min. The resulting suspension was transferred to a glass tube and centrifuged at 3300×g
for 20 min. The lipid phase was removed, evaporated to dryness under a stream of nitrogen and resuspended in 1 ml chloroform:methanol (2:1 by volume). The cell pellet was dissolved in 1 ml of 1 M NaOH and used for the determination of protein. The lipid extracts were subjected to thin layer chromatography on (aluminium sheet supported) silica gel plates. The plates were devel-oped with a solvent of hexane:isopropanol:formic acid (80:30:2 by volume). The nonesterified (i.e. ‘free’) and the esterified cholesterol spots were visualized by iodine vapour, cut out and counted in a scintillation counter. The data were expressed as nmol of [14C]-acetate incor-porated per h and per mg of total cell protein. To analyze the incorporation of acetate into nonsaponifi-able sterols, 0.5 ml of the n-hexane:isopropanol extract were evaporated to dryness and saponified with 3 ml of 15% (wt/vol) KOH in 70% (vol/vol) ethanol at 70°C for 5 h. After adding 6 ml of bidistilled water, the nonsa-ponifiable lipids were extracted with diethyl ether (three times with 6 ml each). The lipid extracts were com-bined, evaporated to dryness and suspended in 5 ml aceton:ethanol (1:1 by volume). One hundred
mi-crolitres of a 1 g/l cholesterol solution (in aceton) and 50 ml of 10% (vol/vol) acetic acid were added and the sterols were precipitated overnight with 2 ml of 0.5% (wt/vol) digitonin, dissolved in 50% (vol/vol) ethanol. After centrifugation (3000×g, 20 min), the precipitate was washed once with 6 ml of aceton:diethylether (1:2 by volume) and once with diethylether alone, dissolved in 1 ml of chloroforin:methanol (2:1 by volume) and subjected to thin layer chromatography as described above. The cholesterol spot was then again counted in a liquid scintillation counter.
2.5. Microsomal HMG-CoA reduclase acti6ity
HMG-CoA reductase activity was determined essen-tially as described [24]. The cells were washed twice with 150 mM NaCl, 50 mM Tris – HCl, pH 7.4, scraped with a rubber policeman and pelleted by centrifugation (900×g, 5 min, 4°C). The cell pellet was kept in liquid nitrogen until the time of assay. Cell extracts were prepared by incubating the pellets at 37°C for 10 min with 5 0 mM K2HPO4, pH 7.4, 5 mM dithiothreitol, 5 mM EDTA · Na2, 0.2 mM KCl, 1% (vol/vol) Triton X-100. The detergent solubilized extract was cen-trifuged at 1000×g for 5 min. Aliquots of the super-nate were assayed for protein [25] and for HMG-CoA reductase activity using [14
C]-HMG-CoA as substrate [24].
2.6. HMG CoA synthase acti6ity
Cytosolic HMG-CoA synthase was partially purified from chicken liver using the method of Gil and cowork-ers [26] with the modifications described previously [19]. The enzyme activity was measured by following the formation HMG-CoA from radioactively labelled ace-tyl-CoA (Ac-CoA) and acetoaceace-tyl-CoA (AcAc-CoA). HMG-CoA was isolated from the reaction mixture by reversed-phase ion-pair chromatography [19].
2.7. Uptake and degradation of [125I]-labelled lipoproteins
We used the procedures described by Goldstein et al [24] with slight modifications [27]. Cells were grown to 70% confluence in RPMI 1640 medium with 10% (vol/
vol) FBS in 24-well tissue culture plates. Cells were pre-incubated for 40 h in medium containing 10% (vol/vol) LPDS to up-regulate LDL receptors. To de-termine cellular uptake (surface binding plus internal-ization) and degradation, cells were incubated for 4 h at 37°C with [125I]-labelled LDL in RPMI 1640 (pH 7.4) in the absence of serum. The amount of [125
I]-la-belled trichloroacetic acid-soluble (noniodide) material in the conditioned medium [24].
2.8. Competiti6e enzyme immunoassayfor LDL receptors
Immunoreactive LDL receptors were measured using a competitive enzyme immunoassay. The LDL receptor was partially purified from bovine adrenals by DEAE cellulose chromatography of octyl-b-D-glucoside
solubi-lized membrane extracts as described [28,29]. LDL re-ceptor containing column fractions were identified by SDS polyacrylamide gradient gel electrophoresis (T=
5 – 12.5%) and immunoblotting with the LDL receptor specific monoclonal antibody C7 [30,31]. The receptor containing DEAE cellulose fractions were adjusted to a protein concentration of 7.5 mg/l and supplemented with octyl-b-D-glucoside and N-hydroxysuccinimide to
yield final concentrations of 80 and 0.5 mM, respec-tively. To immobilize the receptors, 50ml of this solu-tion were pipetted into the wells of CovaLink microplates. To each well, 50ml of 1.0 mM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide were added and the plates were incubated overnight at room tempera-ture. The plates were washed twice with 200 ml PBS (0.14 M NaCl, 2.7 mM KCl, 10 mM Na2HPO4,1.5 mM KH2PO4, pH 7.4) and blocked with 5 g/l casein (in PBS) for at least 2 h. To prepare sample membrane fractions, the cells were washed twice with ice-cold 0.15 M NaCl. The cells were then overlayered with 4 ml ice-cold PBS containing, in addition, 1 mM phenylmethylsulfonylfiu-oride (PMSF), 2.5 mM leupeptin, and 200.000 KIE/I aprotinin, and scraped with a rubber policeman. The cell suspension was centrifuged for 5 min at 1000×g. The resulting pellet was resuspended in 0.5 ml of 50 mM Tris-(hydroxy-methyl)-amino-methane-maleate buffer, pH 6.0, 1 mM PMSF, 2.5 mM leupeptin and 200.000 KIE/l aprotinin. Another 0.5 ml aliquot of the Tris-maleate buffer and octyl-b-D-glucoside to yield a final concentration of 40 mM were added. The suspen-sion was incubated for 45 min on ice and then ultracen-trifuged at 100 000×g. Protein in the supernate membrane fraction was determined according to Lowry [23] as modified by Beisiegel [30].
The membrane specimens were pre-incubated with the LDL receptor specific antibody C7 diluted 1:2000 in PBS containing either 0.05% (vol/vol) Tween 20 or 40 mM octyl-b-D-glucoside plus 0.5 g/l casein overnight at
different concentrations of total protein. One hundred microlitres of these mixtures were loaded in duplicates to the LDL receptor-coated microplate wells and incu-bated for 2 h. The plates were washed three times with PBS-Tween and incubated for 1 h with biotinylated goat anti-mouse IgG (diluted 1:2000) in PBS-Tween 20 plus 0.5 g/1 casein. The wells were washed three times and incubated for 1 h with the ABC Peroxidase Elite
reagent. After three additional washing steps, colour was developed with o-phenylenediamine (9.25 mM in 100 mM sodium citrate, pH 5.0) and H202(1.8 mM) for 15 to 30 min. The reaction was stopped by adding 0.5 M H2SO4and absorbance was read at 450 mn (Titertek MCC 340, Flow Laboratories). Standard curves were obtained by running dilutions of a reference tion of partially purified LDL receptor. This prepara-tion was arbitrarily assumed to contain 100 units of LDL receptors per mg total protein.
2.9. Other methods
Reaction rates were fitted to one-ligand Michaelis and Menten type equations using P.FIT, a non-linear curve fitting program from FIG.P Software (Durham, NC).
3. Results
3.1. Effects of lifibrol on the synthesis of sterols from radioacti6ely labelled precursors
We analysed the effects of lifibrol on the biosynthesis of sterols from [14
C]-acetate in the human hepatoma cell line HepG2. To up-regulate the mevalonate pathway, the cells were pre-incubated with medium containing LPDS. As shown in Fig. 1, lifibrol reduced the produc-tion of sterols in a dose-dependent fashion, regardless whether we considered the production of nonesterified cholesterol, of esterified cholesterol or of nonsaponifi-able sterols. At concentrations of 10−6 and 10−5 M, lifibrol reduced the sterol biosynthesis by approximately 20 and 30%, respectively. In contrast to lifibrol, lovas-tatin strongly inhibited sterol biosynthesis; less than 2×10−8 M lovastatin was sufficient to produce a 20% reduction, 50% reduction was obtained at 2.5×10−8 M, and 10−6
M lovastatin decreased the cholesterol synthesis rate by 80%. This is in perfect agreement with data showing that lovastatin inhibited sterol synthesis from acetate with an IC50 of 2×10
−8 mol
/1 in mouse L-M cells [32]. Lovastatin is a competitive inhibitor of HMG-CoA reductase. We reasoned, if lifibrol also acted directly on one of the rate-limiting steps of the sterol synthesis pathway, then the time kinetics of cholesterol synthesis inhibition by lifibrol and lovastatin should be similar. To test this hypothesis, HepG2 cells were pre-incubated with LPDS containing medium and then received lifibrol or lovastatin for different periods of time at concentrations of 10−5 or 10−6 M, respectively. As predicted from the previous experiment, lifibrol was a less effective inhibitor of the formation of cholesterol (esterified and nones-terified) from [14
parallel, suggesting that lifibrol, much as lovastatin, inhibited the cholesterol synthesis pathway directly rather than by reducing the de novo synthesis or by enhancing the catabolism of a rate-limiting enzyme (Fig. 2).
To identify precisely the step at which lifibrol inhib-ited cholesterol synthesis, we examined its effects on the formation of nonesterified cholesterol from either [14 C]-acetate or [14
C]-mevalonate. As shown in Fig. 3, both lifibrol and lovastatin decreased the synthesis of choles-terol from acetate but not from mevalonate, indicating that lifibrol inhibited the production of mevalonate rather than its conversion to sterols.
The production of mevalonate is catalyzed by two sequentially acting enzymes, cytosolic HMG-CoA syn-thase and microsomal HMG-CoA reductase. Cytosolic HMG-CoA synthase is responsible for the generation of CoA from Ac-CoA and AcAc-CoA. HMG-CoA reductase converts HMG-HMG-CoA to mevalonate. We first examined whether lifibrol influenced microsomal HMG-CoA reductase. HepG2 cells were pre-incubated with medium containing LPDS. The cells were dis-rupted and HMG-CoA reductase in the homogenate
Fig. 2. Effect of lifibrol and lovastatin on the incorporation of acetate intode no6osynthesized cellular sterols. HepG2 cells were grown in RPMI 1640 medium supplemented with 10% (vol/vol) FBS. Forty hours prior to the experiment, the cells were switched to medium containing 10% (vol/vol) human LPDS. The cells were then incubated with lifibrol (circles) or lovastatin (squares) at final concentrations of 10−5or 10−6M, respectively, or in the absence of drugs for the time
intervals indicated on the abscissa. One hour before the end of each incubation period, the cells were pulse-labelled with [14C]-acetate
(specific activity 56 mCi/mmol, final concentration 35 mM) and the incorporation of acetate into esterified (left panel) and nonesterified (right panel) cholesterol was determined as described in Section 2. [3H]-cholesterol was used as an internal standard for the recovery of
cholesterol. Results are normalized to cellular protein and expressed in percent of control incubations performed for the respective time periods in the absence of drugs. The data are means from duplicates; the standard deviations of the replicates were 11% or less of the respective means. The average 100% of control rates of [14C]-acetate
incorporations into nonesterified cholesterol and into esterified cholesterol were 6.5 and 0.7 nmol/h/mg, respectively.
Fig. 1. Effect of lifibrol and lovastatin on the incorporation of acetate intode no6osynthesized cellular sterols. HepG2 cells were grown in RPMI 1640 medium supplemented with 10% (vol/vol) FBS. Forty hours prior to the experiment, the cells were switched to medium containing 10% (vol/vol) human LPDS. The cells then received lifibrol (left panel) or lovastatin (right panel) at the concentrations indicated on the abscissa for 8 h. Two hours before the end of the incubation period, the cells were pulse-labelled with [14C]-acetic acid,
sodium salt (specific activity 56 mCi/mmol, final concentration 35 mM). The incorporation of acetate into esterified (circles) and nones-terified (squares) cholesterol or into the digitonin precipitable sterols (triangles) were determined as described in Section 2. [3H]-cholesterol
was used as an internal standard for the recovery of cholesterol. Results were normalized to the amount of cellular protein and expressed in percent of the respective control incubations. The data are means from duplicates, except that the controls are average values of four replicate incubations. The standard deviations of the repli-cates were 9% or less of the respective means. The average 100% of control rates of [14C]-acetate incorporations into esterified (circles),
nonesterified (squares) cholesterol, and into digitonin precipitable sterols (triangles) incorporations were 0.76, 4.11, and 3.78 nmol/h/ mg, respectively.
was measured in the presence of increasing concentra-tions of lifibrol and lovastatin. As shown in Fig. 4, lovastatin dose-dependently inhibited HMG-CoA re-ductase, the IC50 being around 3×10−8 M. In con-trast, lifibrol did not affect HMG-CoA reductase. We then examined the effect of lifibrol on HMG-CoA synthase using partially purified enzyme from chicken liver and a reversed-phase ion-pair chromatography based method to recover HMG-CoA, the product of the reaction, from the incubation mixture [19]. Lifibrol (10−5M) inhibited HMG-CoA synthase (Fig. 4). When the concentration of AcAc-CoA was kept constant at 60 mM, the inhibitory effect of lifibrol was greatest between 400 and 1200 mM Ac-CoA. Further increases of the Ac-CoA substrate concentration reversed the inhibitory effect of lifibrol; at 4000 mM Ac-CoA, HMG-CoA synthase activity was inhibited by 10% only. To determine the type of inhibition, we estimated
3.2. Effects of lifibrol on the acti6ity of the LDL
receptor
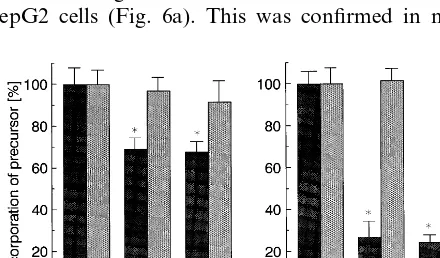
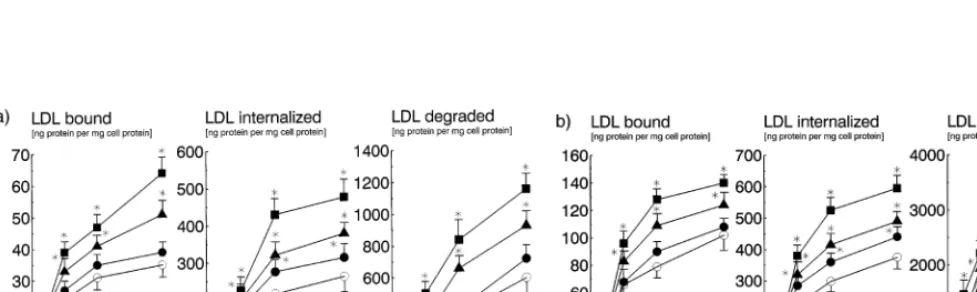
To examine the effect of lifibrol on the activity of LDL receptors, we measured receptor mediated uptake and degradation of [125I]-1abelled LDL in HepG2 cells and normal human skin fibroblasts. The cells were incubated for 40 h with medium containing LPDS alone or with the same medium containing, in addition, lifibrol at concentrations of 10−7– 10−5 M. As shown in Fig. 5a, lifibrol increased receptor mediated endocy-tosis of LDL in both cell types in a dose-dependent fashion. At 10−5
M, lifibrol increased receptor medi-ated binding, uptake, and degradation by approxi-mately 50% in human skin fibroblasts (Fig. 5b). The effect of lifibrol was even greater in HepG2 cells, where lifibrol enhanced receptor mediated catabolism of LDL by :80%. We wished to compare the effects of lifibrol
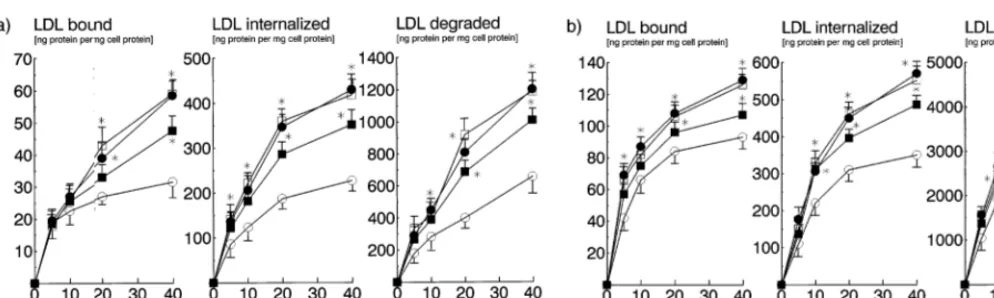
and lovastatin on the receptor mediated uptake of LDL. In these studies, lifibrol and lovastatin were used at 10−5 and 10−6 M, respectively. At these concentra-tions, the two compounds inhibited sterol synthesis by 30 and 80%, respectively. However, despite the smaller inhibition of sterol synthesis, 10−5
M lifibrol was more effective in enhancing receptor mediated binding, up-take, and degradation of LDL than lovastatin in HepG2 cells (Fig. 6a). This was confirmed in normal
Fig. 4. Effects of lifibrol and lovastatin on the activity of microsomal HMG-CoA reductase and HMG-CoA synthase. Left panel:HepG2 cells were grown in 80 cm2polystyrene flasks containing RPMI 1640
medium supplemented with 10% (vol/vol) FBS. To upregulate HMG-CoA reductase, the cells were incubated with medium containing 10% (vol/vol) human LPDS for 48 h before the experiment. Cell extracts were prepared by lysis with 50 mM potassium phosphate, pH 7.4, 5 mM dithiothreitol, 5 mM EDTA, 200 mM KCl, 1% (vol/vol) Triton X-100, and pre-incubated at 38°C for 10 min with lifibrol (circles) or lovastatin (squares) at the indicated concentrations. The extracts were then assayed for HMG-CoA reductase activity as described in Section 2 using [3H]-mevalonate as an internal standard. Two independent
experiments were performed with Lifibrol (open and closed circles, respectively). The results are means from duplicates, normalized to extract protein. The 100% of control values for HMG-CoA reductase activity were: 158 (lifibrol, closed circles), 184 (lifibrol, open circles), and 106 (lovastatin, squares) pmol/h/mg, respectively. Right panel: cytosolic HMG-CoA synthase was purified from chicken liver as described [19,26]. The activity of the partially purified enzyme was determined using 60mM AcAc-CoA and the indicated concentrations of [1-14C]-Ac-CoA as substrates, either in the absence (circles) or in
the presence of Lifibrol (10−5 M, squares). HMG-CoA synthase
activity was measured using an assay in which the HMG-CoA formed during the reaction is isolated by reversed phase-ion pair chromatog-raphy [19].
Fig. 3. Effect of lifibrol and lovastatin on the incorporation of acetate or mevalonate into nonesterified cholesterol. HepG2 cells were grown in RPMI 1640 medium supplemented with 10% (vol/vol) FBS. Forty hours prior to the experiment, the cells were switched to medium containing 10% (vol/vol) human LPDS. The cells were then incubated with lifibrol (left panel) or lovastatin (right panel) at the indicated concentrations for 8 h. Two hours before the end of the incubation period, the cells were pulse-labelled with either [14C]-acetate (solid
bars) or [14C]-mevalonate (grey bars, both specific activity 56 mCi/
mmol, final concentration 3 5 mM). The rates of incorporation of radioactively labelled precursors into nonesterified cholesterol was determined after separating the cellular lipids by thin layer chro-matography. [3H]-Cholesterol was included as an internal standard to
correct for the recovery of cholesterol. The data are means from two experiments, each performed in duplicate. Results are normalized to cellular protein and expressed in percent of the control incubations. *: PB0.05 versus the respective control incubations. The average rates of [14C]-acetate and [14C]-mevalonate incorporations in the control
incubations were 3.48 and 0.65 nmol/h/mg, respectively.
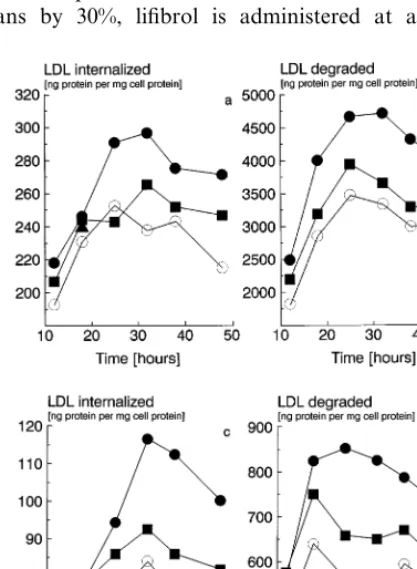
human skin fibroblasts (Fig. 6b). No additional effect was obtained when lifibrol and lovastatin were used in combination. To determine whether upregulation of LDL receptors by lovastatin and lifibrol followed dif-ferent time kinetics, we incubated human skin fibrob-lasts with medium containing LPDS plus lifibrol or lovastatin for time intervals between 12 and 48 h. Compared to medium containing LPDS alone, both drugs enhanced LDL receptor mediated endocytosis (Fig. 7a and b). Interestingly, the effect of lifibrol occurred earlier and was stronger than that of lovastatin.
lova-statin in FBS containing medium which provides cholesterol sufficient to suppress LDL receptors in the absence of drugs. Under these conditions, lifibrol still markedly enhanced receptor mediated uptake and degradation of radiolabelled LDL at all incubation times considered (Fig. 7c and d).
To analyse the effects of lifibrol on the relative amount of immunoreactive LDL receptors of human skin fibroblasts, we developed a competitive enzyme immunoassay. Partially purified LDL receptors were immobilized to microwell plates. Cell membrane sam-ples were incubated with the LDL receptor specific monoclonal antibody C7, either in the presence of octyl-b-D-glucoside (40 mM) or Tween 20 (0.5 mM) as detergent. Antibody not absorbed to LDL receptors in the sample was then allowed to bind to the immobilized bovine receptors and quantitated. The assay was cali-brated using a preparation of bovine adrenal LDL receptors. This preparation was arbitrarily considered to contain 100 units of LDL receptor per mg protein. As expected, we obtained an inverse relationship be-tween the amount of LDL receptors in the sample and the absorbances (Fig. 8). The presence of octyl-b-D
-glu-coside during incubation of the membrane extracts with the monoclonal antibody C7 improved the immunore-activity of the LDL receptors compared to Tween 20 (Fig. 8). When we estimated LDL receptors in mem-branes from cells pre-incubated with lifibol (10−5 M) approximately 2-fold increases over control cells was observed. In contrast, lovastatin (10−6 M) enhanced the amount of immunoreactive LDL receptors by not more than 1.5-fold. These data indicate that the en-hanced uptake and degradation caused by lifibrol was
paralleled by an increase of the number of receptor molecules on the cell surface (Fig. 8).
3.3. Effect of lifibrol on the expression of HMG-CoA reductase in cultured cells
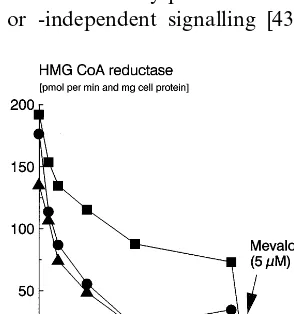
The expression of HMG-CoA reductase in cultured cells is regulated by sterol and non-sterol mevalonate derived products [33]. In cells incubated with HMG-CoA reductase inhibitors at a concentration sufficient to block the production of the nonsterol mevalonate derived effectors of HMG-CoA reductase, extracellular cholesterol only partly suppresses HMG-CoA reduc-tase. We wondered whether lifibrol, similarly to HMG-CoA reductase inhibitors, affected the provision of cells with the regulatory mevalonate derived products. HepG2 cells were incubated in medium containing LPDS alone or, in addition, lifibrol or lovastatin and increasing concentrations of LDL as a source of extra-cellular cholesterol. After washing the monolayers three times with 3 ml of 50 mM Tris – HCl, 150 mM NaCl, pH 7.4, to remove the drugs, we determined the activity of the microsomal HMG-CoA reductase in the cells. In the absence of extracellular LDL, both lifibrol and lovastatin stimulated HMG-CoA reductase at approxi-mately the same rate over lipoprotein deficient medium alone (Fig. 9). However, the two compounds behaved entirely differently when exogenous LDL was added at increasing concentrations. Whereas the lifibrol induced increase in HMG-CoA reductase could completely be repressed to control levels by LDL, mevalonate plus LDL was required to suppress the lovastatin mediated increase in reductase activity.
Fig. 5. Effect of lifibrol on the uptake and degradation of LDL in cultured cells. HepG2 cells (panel 5a) and human skin fibroblasts (panel 5b) were grown in RPM1 1640 medium supplemented with 10% (vol/vol) FBS. The cells were incubated for 40 h with medium containing 10% (vol/vol) LPDS alone (open circles) or with Lifibrol at concentrations of 10−7M (circles), 10−6M (triangles) or 10−5M (squares), respectively.
The cells then received [125I]-1abelled LDL (1.030BdB1.050 kg/l) at the indicated concentrations. Binding at CC (left panels), uptake (center panels), and degradation (right panels) at 37°C of [125I]-labelled LDL were determined as described in Section 2. Each data point represents the
Fig. 6. Effect of lifibrol and lovastatin on the uptake and degradation of LDL in cultured cells. HepG2 cells (panel 6a) and human skin fibroblasts (panel 6b) were grown in RPMI 1640 medium supplemented with 10% (vol/vol) FBS. The cells were incubated for 40 h with medium containing 10% (vol/vol) LPDS alone (open circles) or, in addition, Lifibrol (10−5M, solid circles), lovastatin (10−6M, squares) or the combination of both
lifibrol and lovastatin (10−5and 10−6M, respectively, open squares). The cells then received [125I]-labelled LDL at the indicated concentrations.
Binding at 4°C (left panels), uptake (center panels) and degradation at 37°C (right panels) of [125I]-LDL (1.030 – 1.050 kgl) were measured as
described in Section 2. Each data point represents the average from two independent experiments, each performed in triplicate. Data are adjusted for non-specific binding, uptake, and degradation determined in the presence of a 50-fold excess of unlabelled LDL. *P B 0.05 versus the respective control incubations.
4. Discussion
Lifibrol is a novel, effective lipid lowering drug. In rats, marmosets, pigs, and in humans, it decreases cholesterol and LDL cholesterol [11 – 15]. The aim of this study was to characterize the effects of lifibrol on the metabolism of cholesterol in cultured cells. Cellular cholesterol originates from two sources, the de novo synthesis from activated acetic acid and the receptor mediated uptake of cholesterol containing lipoproteins. These two pathways are delicately balanced. They con-trol both the intracellular level of cholesterol and the concentration of cholesterol transporting lipoproteins in the circulation. Here we present strong evidence that lifibrol exerts independent but synergistic effects on both of the two pathways, cholesterol biosynthesis and uptake, which may explain efficacy of lifibrol as a cholesterol lowering agent in vivo.
In HepG2 cells, therapeutic concentrations of lifibrol inhibited the synthesis of sterols from acetic acid but not from mevalonate, indicating that it acted on one of the mevalonate generating steps of the sterol synthesis pathway. The synthesis of mevalonate is catalyzed by two sequential enzymes, cytosolic HMG-CoA synthase and microsomal HMG-CoA reductase. Lifibrol com-petitively inhibited HMG-CoA synthase, but not HMG-CoA reductase. The inhibition constant Ki of lifibrol, however, was 108mM compared to a Michaelis constant Km of 1408 mM for the AcCoA substrate of HMG-CoA synthase. Lifibrol is thus far less effective in inhibiting HMG-CoA synthase than lovastatin in in-hibiting HMG-CoA reductase (inhibition constant Ki, 6.4×10−10
M; Michaelis constant Km, 4×10
−6 M [34]). Consistently, lifibrol and lovastatin reduced the sterol synthesis from acetic acid in cultured cells at
significantly different degrees (20 and 80%, respectively, at concentrations of 10−6 M).
In humans, cholesterol lowering by lifibrol is at least as strong and occurs more rapidly than by HMG-CoA reductase inhibitors [13,14]. We, therefore, hypothe-sized that the inhibition of cholesterol synthesis ob-served in cell culture was not sufficient to account for the marked effects of lifibrol in6i6o.
As LDL receptors are a major determinant of the LDL concentration [6,35], we examined the effect of lifibrol on the activity of LDL receptors in cell culture. Since the HMG-CoA reductase inhibitors lower choles-terol by stimulating the receptor mediated catabolism of LDL [10,36,37], we also wished to compare the effects of lifibrol and lovastatin. Under the condition of cholesterol starvation, i.e. in lipoprotein deficient medium, 10−6 M lovastatin enhanced the receptor mediated uptake of LDL by approximately 20% over the level reached by LPDS alone. This data is in good agreement with previous studies in fibroblasts in which 2.6×10−6M compactin enhanced LDL receptor activ-ity by approximately 25% in human skin fibroblasts [38]. Lifibrol stimulated the receptor mediated uptake of LDL in a dose dependent manner. At a concentra-tion of 10−6 M lifibrol and lovastatin were approxi-mately equally effective. In the subsequent studies we used lifibrol and lovastatin at concentrations of 10−5 M and 10−6
M, respectively. These conditions were chosen for the following reasons. As shown above, the two compounds inhibited sterol synthesis at signifi-cantly different rates. To compare the two drugs at doses equally effective with regard to sterol synthesis inhibition, we had two alternatives. The first was to use lovastatin at 2×10−8
how-ever, that this lovastatin concentration was too low to result in measurable effects on the LDL receptor. We, therefore, decided to use lovastatin at 10−6M which is a concentration known to elicit measurable stimulation of LDL receptors. At 10−6 M, lovastatin inhibits cholesterol synthesis by 80%. To produce a similar inhibition of sterol synthesis more than 10−4M lifibrol would have been required. As assessed by studies of [3H]-thymidine and [14C]-alanine incorporation concen-trations exceeding 10−4M lifibrol were cytotoxic (data not shown). To avoid non-specific cytotoxic effects, we decided to use lifibrol at 10−5
M along with 10−6 M lovastatin although inhibition of cholesterol synthesis was different at these concentrations (30 and 80%, respectively). Using lifibrol at a 10-fold higher concen-tration than lovastatin is, in addition, consistent with clinical experience. To lower LDL cholesterol in hu-mans by 30%, lifibrol is administered at a dose of
Fig. 8. Effect of Lifibrol on the expression of immunoreactive fibrob-last LDL receptors. Normal human skin fibrobfibrob-lasts were grown in RPMI 1640 medium supplemented with 10% (vol/vol) FBS. The cells were incubated for 36 h with medium containing 10% (vol/vol) LPDS alone (triangles) or, in addition, lifibrol (10−5M, circles) or
lovas-tatin (10−6 M, squares). At the end of the incubation period, cell
membranes were prepared and adjusted to the total protein concen-trations indicated on the abscissas. LDL receptors were determined using a solid phase competitive enzyme immunoassay based on the LDL receptor specific monoclonal antibody C7 as described in Sec-tion 2. A standard curve was constructed with a reference preparaSec-tion of LDL receptors partially purified from bovine adrenal membranes (inverted triangles) which was considered to contain 100 units of LDL receptors per mg total protein. Each data point represents the average from duplicates. Left panel: incubation of the membrane extracts with the monoclonal antibody C7 in PBS containing 0.05% (vol/vol) Tween 20 and 0.05% (wt/vol) casein. Using only those values below and above the lowest and the highest value of the standard curve, respectively (solid symbols), the relative content of LDL receptors of the fibroblast membrane preparations was calculated to be, on aver-age, 9.7 units and 18.7 units/mg protein in the cells incubated with LPDS and in the cells with LPDS plus lifibrol, respectively. Right panel: incubation of the membrane extracts with the monoclonal antibody C7 in PBS containing 40 MM octyl-b-D-glucoside and
0.05% (wt/vol) casein. Using the values indicated by the solid sym-bols, the relative content of LDL receptors of the fibroblast mem-brane preparations was calculated to be, on average 12.4, 18.0 and 24.8 units/mg membrane protein in the cells incubated with LPDS, LPDS plus lovastatin, or LPDS plus Lifibrol, respectively.
Fig. 7. Effect of lifibrol on the uptake and degradation of LDL in human skin fibroblasts: Time kinetics. Normal human skin fibroblasts were grown in RPMI 1640 medium supplemented with 10% (vol/vol) FBS. The cells were incubated for the time intervals indicated on the abscissa and then received [125I] LDL (30 mg/1 protein, 1.030BdB
1.050 kg/1) for another 4 h. Panels a and b (top): medium containing 10% (vol/vol) LPDS; panels c and d (bottom): medium containing 10% (vol/vol) FBS. Open circles: medium without drugs; squares: lovastatin (10−6M); circles: lifibrol (10−5M). Uptake (left panels, a
and c) and degradation (right panels, b and d) were determined as described in Section 2. Each data point represents the average from triplicates. The standard deviations of the replicates were 10% or less of the respective means. Data are adjusted for non-specific uptake, and degradation determined in the presence of a 50-fold excess of unlabelled LDL.
able to overcome downregulation of LDL receptors by sterols or, alternatively, renders the cell less responsive to feedback regulation by raising the threshold level of cholesterol in the regulatory pool at which LDL recep-tor activity and RMG-CoA reductase are suppressed. The increase in LDL receptor mediated endocytosis by lifibrol was accompanied by increases in the amounts of the immunologically detectable LDL recep-tors, indicating that lifibrol enhanced the production of LDL receptors. Instable isotope kinetics with patients with hyperlipoproteinemia, lifibrol treatment resulted in an accelerated LDL apoB metabolism with decreased LDL cholesterol levels. This was mainly due to an increased LDL catabolism, which was partially attenu-ated by an increase in LDL production [20]. The precise molecular mechanism underlying this action of lifibrol remains to be determined. In recent years, evidence has accumulated that sterol independent pathways are in-volved in the regulation of LDL receptors. Insulin stimulates high affinity binding of LDL in a variety of cell types in culture [39,40], even when LDL is present in the medium [41]. Mitogenic factors like platelet derived growth factor [42,43], epidermal growth factor [44,45] or oncostatin M [44,46] increase LDL receptor activity, mediated either by protein kinase C-dependent [44,47,48] or -independent signalling [43,45]. Whether
or not the stimulation of LDL receptors in response to estrogens [49] is secondary to a depletion of the regula-tory cholesterol pool [50] remains to be settled. Finally, Hu¨ttinger et al. showed that HOE-402, another potent lipid lowering agent, utilizes a sterol-independent path-way to activate LDL receptorsin6itroandin6i6o [51]. The question whether one or the other of these non-sterol regulatory processes is affected by lifibrol is currently under investigation in our laboratories.
When incubated with HepG2 cells in the absence of extracellular cholesterol, lifibrol and lovastatin stimu-lated HMG-CoA reductase activity to almost the same extent. The expression of HMG-CoA reductase is un-der multivalent feedback control [33]. In the presence of HMG-CoA reductase inhibitors, exogenous LDL was unable to suppress HMG-CoA reductase completely. However, if mevalonate is supplied together with LDL, complete down-regulation is achieved, confirming the previous observation that the expression of HMG-CoA reductase is regulated by both sterols and non-sterol compounds derived from mevalonate [33]. Sterol medi-ated control of reductase expression is exerted at the transcriptional level; non-sterol mediated regulation resides at the post-transcriptional level [52 – 54]. The increase in HMG-CoA reductase brought about by lifibrol was completely suppressed by LDL, suggesting a transcriptional rather than a post-transcriptional mechanism of induction. Most importantly, however, these data indicate that lifibrol does not prevent the synthesis of regulatory active non-sterol mevalonate derived compounds.
In conclusion, we demonstrate that the mechanism of action of lifibrol is distinct from that of the most widely used lipid lowering drugs, the HMG-CoA reductase inhibitors. We predict from our data that the strong cholesterol lowering effect of lifibrol in vivo results from its stimulatory effect on LDL receptors. The inhibition of cholesterol synthesis observed in vitro appears too weak to account per se for the in vivo effects of lifibrol. This may turn out a major advantage as lifibrol would enhance the removal of cholesterol from the circulation without affecting the provision of biologically essential products — ubiquinone, farnesy-lated proteins — of the mevalonate pathway.
Acknowledgements
This work was supported by a research grant from Klinge Pharma GmbH, Munich (Germany). The au-thors thank Dr Manfred Hu¨ttinger, Institute of Medi-cal Chemistry, Vienna, and Professor Klaus Seibel, Klinge Phanna GmbH, for discussion; Dr Michael Hoffmann for critical reading of the manuscript; Dr Manfred W. Baumstark, Institute of Sports Medicine, Freiburg, and Dr Horst Mayer, Central Laboratory,
Fig. 9. Effect of lifibrol and lovastatin on cholesterol mediated suppression of LINIG-CoA reductase in HepG2 cells. HepG2 cells were grown in six well cell culture plates in RPM1 1640 medium supplemented with 10% (vol/vol) FBS and used at a confluence of approximately 70%. The cells were then incubated for 18 h in medium containing 10% (vol/vol) human LPDS and LDL at the concentra-tions indicated on the abscissa, either alone (triangles) or, in addition, containing lifibrol (10−5M, circles) or lovastatin (10−6M, squares),
respectively. To the one set of incubations containing the highest concentration of LDL and 10−6M lovastatin, mevalonate was added
at a final concentration of 5 mM (indicated by the arrow). Microso-mal HMG-CoA reductase activity was determined in cell extracts as described in Section 2 using [3H]-mevalonate as an internal standard.
General Hospital KarIsruhe, for help with the non-lin-ear curve fitting.
References
[1] Sacks FM, Keffer MA, Moye LA, et al. The Cholesterol and Recurrent Events Trial Investigators: the effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. New Engl J Med 1996;335:1001 – 9. [2] The Long-Term Intervention with Pravastatin in Ischaemic
Dis-ease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. New Engl J Med 1998;339:1349 – 57.
[3] Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS — Air Force/Texas Coronary Atherosclerosis Prevention Study. J Am Med Assoc 1998;279:1615 – 22.
[4] Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease — The Scandinavian Simvastatin Survival Study (4S). Lancet 1994;344:1383 – 9.
[5] West of Scotland Coronary Prevention Study Group, Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolernia. New Engl J Med 1995;333:1301 – 7.
[6] Brown MS, Goldstein JL. A receptor mediated pathway for cholesterol homeostasis. Science 1986;232:34 – 47.
[7] Soutar AK, Myant NB, Thompson GR. The metabolism of very low density and intermediate density lipoproteins in patients with familial hypercholesterolemia. Atherosclerosis 1982;43:217 – 31.
[8] Goldstein JL, Brown MS. Regulation of the mevalonate path-way. Nature 1990;343:425 – 30.
[9] Endo A, Kuroda M, TsuJita Y. 236A, 236B and ML-236C, new inhibitors of cholesterogenesis produced by Penicil-lium citrinum. J Antibiot 1976;29:1346 – 8.
[10] Ma PTS, Gil G, Stidhof TC, et al. Mevinolin, an inhibitor of cholesterol synthesis, induces mRNA for low density lipoprotein receptor in livers of hamsters and rabbits. Proc Natl Acad Sci USA 1986;83:8370 – 4.
[11] Schliack M, Loser R, Seibel H, et al. Hypolipidemic activity of K12.148 in rats, marmosets and pigs. Artery 1989;16:90 – 104. [12] Schliack M, Loser R, Seibel K, et al. The hypolipidemic effect of
Lifibrol during a long term treatment of pigs. Artery 1990;18:1 – 15.
[13] Hasibeder H, Staab HJ, Seibel K, et al. Clinical pharmacology of the hypocholesterolemic agent K12.148 (Lifibrol) in healthy vol-unteers. Eur J Clin Pharmacol 1991;40:S91 – 4.
[14] Schwandt P, Elsasser R, Schmidt C, et al. Safety and efficacy of lifibrol upon four-week administration to patients with primary hypercholesterolemia. Eur J Clin Pharmacol 1994;47:133 – 8. [15] Lifibrol Study Group, Locker PK, JungbIuth GL, Francom SF,
et al. Lifibrol: a novel lipid – lowering drug for the therapy of hypercholesterolemia. Clin Pharmacol Ther 1994;57:73 – 88. [16] Grundy SM. HMG – CoA reductase inhibitors for treatment of
hypercholesterolemia. New Engl J Med 1988;319:24 – 32. [17] Todd PA, Goa KL. Simvastatin: a review of its pharmacological
properties and therapeutic potential in hypercholesterolemia. Drugs 1990;40:583 – 607.
[18] McTavish D, Sorkin EM. Pravastatin: a review of its pharmaco-logical properties and therapeutic potential in hypercholesterol-ernia. Drugs 1991;42:65 – 89.
[19] ScharnagI H, Ma¨rz W, Schliack H, et al. A novel assay for cytosolic 3-hydroxy-3-methylglutaryl-coenzyme A synthase activ-ity using reversed-phase ion-pair chromatography. J Lipid Res 1995;36:622 – 7.
[20] Winkler K, Schafer JR, Klima B, et al. Lifibrol enhances the low density ipoprotein apolipoprotein B- 100 turnover in patients with hypercholesterolemia and mixed hyperlipidemia. Atherosclerosis 1999;144:167 – 75.
[21] Cutts JL, Meirtykovych G. Defective utilization of cholesterol esters from low-density lipoproteins in a human acute lymphoblastic leukemia T cell line. Biochim Biophys Acta 1988;961:65 – 72.
[22] Goldstein JL, Brown MS. Binding and degradation of low density lipoproteins by cultured human fibroblasts: comparison of cells from a normal subject and from a patient with ho-mozygous familial hypercholesterolemia. J Biol Chem 1974;249:5153 – 62.
[23] Lowry OH, Rosebrough NJ, Farr AL, et al. Protein measure-ment with the folin phenol reagent. J Biol Chem 1951;193:265 – 75.
[24] Goldstein, JL, Basu, SK, Brown, MS. Receptor-mediated endo-cytosis of low density lipoprotein in cultured cells. In: Fleischer S, Fleischer B, editors. Biomembranes. Part L. Membrane Bio-genesis. Methods in Enzymology, vol. 98. Orlando: Academic Press. 1983: 241 – 60.
[25] Bensadoun A, Weinstein D. Assay of proteins in the presence of interfering materials. Anal Biochem 1976;70:241 – 50.
[26] Gil G, Goldstein JL, Slaughter CA, et al. Cytoplasmic 3-hy-droxy-3-methylgIutaryl coenzyme A synthase from the hamster. 1. Isolation and sequencing of a full-length cDNA. J Biol Chem 1986;261:3710 – 6.
[27] Ma¨rz W, Baumstark MW, ScharnagI H, et al. Accumulation of ‘small dense’ low density lipoproteins in a homozygous patient with familial defective apolipoprotein B – 100 results from hetero-geneous interaction of LDL-subfractions with the LDL receptor. J Clin Invest 1993;92:2922 – 33.
[28] Schneider WJ, Goldstein JL, Brown MS. Partial purification and characterization of the low density lipoprotein receptor from bovine adrenal cortex. J Biol Chem 1980;255:11442 – 7. [29] Schneider WJ, Basu SK, McPhall MJ, et al. Solubilization of the
low-density lipoprotein receptor. Proc Natl Acad Sci USA 1979;76:5577 – 81.
[30] Beisiegel U, Schneider WJ, Goldstein JL, et al. Monoclonal antibodies to the low density lipoprotein receptor as probes for study of receptor-mediated endocytosis and the genetics of famil-ial hypercholesterolernia. J Biol Chem 1981;256:11923 – 31. [31] Beisiegel U, Schneider WJ, Brown MS, et al. Immunoblot
analy-sis of low density lipoprotein receptors in fibroblasts from sub-jects with familial hypercholesterolemia. J Biol Chem 1982;257:13150 – 6.
[32] Albers-Schonberg G, Joshua H, Lopez MB. Dihydromevinolin, a potent hypocholesterolemic metabolite produced byaspergillus terreus. J Antibiot 1981;34:507 – 12.
[33] Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating iso-prenoid synthesis and cell growth. J Lipid Res 1980;21:505 – 17. [34] Alberts AW, Chen J, Kuron G, et al. Mevinolin: A highly potent competitive inhibitor of hydroxmethylglutaryl-coenzyme A re-ductase and a cholesterol-lowering agent. Proc Natl Acad Sci USA 1980;77:3957 – 61.
[35] Kovanen PT, Bilheimer DW, Goldstein JL, et al. Regulatory role for hepatic low density lipoprotein receptors in vivo in the dog. Proc Natl Acad Sci USA 1981;78:1194 – 8.
[37] Reihner E, Rudeling M, Staffiberg D, et al. Influence of prava-statin, a specific inhibitor of HMG-CoA reductase, on hepatic metabolism of cholesterol. New Engl J Med 1990;323:224 – 8. [38] Brown MS, Faust JR, Goldstein JL, et al. Induction of
3-hy-droxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J Biol Chem 1978;253:1121 – 8. [39] Chait A, Bierman EL, Albers JJ. Low-density lipoprotein
recep-tor activity in cultured human skin fibroblasts: mechanism of insulin-induced stimulation. J Clin Invest 1979;64:1309 – 19. [40] Streicher R, Kotzka J, Mfiller-Wieland D, et al. SREBP-1
medi-ates activation of the low density lipoprotein receptor promoter by insulin and insulin-like growth factor-1. J Biol Chem 1996;271:7128 – 33.
[41] Wade DP, Knight BL, Soutar AK. Hormonal regulation of low-density lipoprotein (LDL) receptor activity in human HepG2 cells. Insulin increases LDL receptor activity and dimin-ishes its suppression by exogenous LDL. Eur J Biochem 1988;174:213 – 8.
[42] Chait A, Ross R, Albers JJ, et al. Platelet-derived growth factor stimulates activity of low density lipoprotein receptors. Proc Natl Acad Sci USA 1980;77:4084 – 8.
[43] Roth M, Emmons LR, Perruchoud A, et al. Expression of the low density lipoprotein receptor and 3-hydroxy-3-methyl coen-zyme A reductase genes are stimulated by recombinant platelet-derived growth factor isomers. Proc Natl Acad Sci USA 1991;88:1888 – 92.
[44] Grove RI, Mazzuco CE, Radka SF, et al. Oncostatin M upregu-lates low density lipoprotein receptors in HepG2 cells by a novel mechanism. J Biol Chem 1991;266:18194 – 9.
[45] Graham A, Russell U. Stimulation of low-density lipoprotein uptake in HepG2 cells by epidermal growth factor via tyrosine kinase-dependent, but protein kinase C-independent, mechanism. Biochem J 1994;298:579 – 84.
[46] Liu J, Streiff R, Zhang YL, et al. Novel mechanism of transcrip-tional activation of hepatic LDL receptor by oncostatin M. J Lipid Res 1997;38:2035 – 48.
[47] Auwerx JH, Chait A, Wolfbauer G, et al. Involvement of second messengers in the regulation of the low density lipoprotein receptor gene. Mol Cell Biol 1989;9:2298 – 302.
[48] Kamps JAAM, van Berkel TX. Regulation of low-density lipo-protein receptors in the human hepatoma cell line HepG2. Eur J Biochem 1993;213:989 – 94.
[49] Ma P, Yamamoto T, Goldstein JL, et al. Increased mRNA for low density lipoprotein receptor in livers of rabbits treated with 17-alpha-ethinyl estradiol. Proc Natl Acad Sci USA 1986;83:792 – 6.
[50] Semenkovich CF, Ostlund RE. Estrogens induce low-density lipoprotein receptor activity and decrease intracellular choles-terol in human hepatoma cell line HepG2. Biochemistry 1987;26:4987 – 92.
[51] Huettinger M, Hermann M, Goldenberg H, et al. Hypolipidemic activity of HOE-402 is mediated by stimulation of the LDL receptor pathway. Arteriosler Thromb 1993;13:1005 – 12. [52] Osborne TF, Gil G, Goldstein JL, et al. Operator constitutive
mutation of 3-hydroxy-3-methylglutaryl coenzyme A reductase abolishes protein binding to sterol regulatory element. J Biol Chem 1988;263:3380 – 7.
[53] Nakanishi M, Goldstein JL, Brown MS. Multivalent control of 3-hydroxy-3-methylgIutaryl coenzyme a reductase. Meval-onate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J Biol Chem 1988;263:8929 – 37.
[54] Hidaka Y, Hotta H, Nagato Y, et al. Effect of a novel squalene epoxidase inhibitor, NB-598, on the regulation of cholesterol metabolism in HepG2 cells. J Biol Chem 1991;266:13171 – 7.