I. PENDAHULUAN A. Latar Belakang
Klasifikasi bakteri secara filogenetik dapat dilakukan melalui analisis taksonomi molekular (Sembiring, 2010). Data yang digunakan adalah sekuens gen yang mengkode 16S rRNA (16S rDNA) pada masing-masing strain yang akan diklasifikasikan. Woese pada tahun 1980-an telah dapat menyimpulkan bahwa perbandingan filogenetik berdasarkan bagian conserved dari genom lebih stabil daripada klasifikasi berdasarkan pada sifat-sifat fenotipik (Woese, 1987). Oleh karena itu, penggunaan molekul rRNA disebarluaskan untuk pembuatan perbandingan filogenetik, dan gen 16S rRNA (1650 bp) adalah marker yang paling umum digunakan dalam bidang sistematika mikrobia (Prakash et al., 2007).
Molekul 16S rRNA terdiri atas daerah variabel dan daerah conserved, dan primer universal untuk amplifikasi gen 16S rRNA biasanya dipilih dari daerah conserved sedangkan daerah variabel digunakan untuk taksonomi perbandingan. Langkah awal dalam klasifikasi filogenetik adalah mengisolasi dan memurnikan DNA kromosomal dari masing-masing strain. Gen 16S RNA selanjutnya diamplifikasi dengan teknik PCR (polymerase chain reaction) dari masing-masing sampel DNA kromosomnya. Hasil amplifikasi tersebut dimurnikan untuk disekuensing (Prakash et al., 2007).
Analisis filogenetika, kelompok outgroup sangat dibutuhkan dan menyebabkan polarisasi karakter atau ciri, yaitu karakter apomorfik dan plesiomorfik. Karakter apomorfik adalah karakter yang berubah dan diturunkan dan terdapat pada ingroup, sedangkan karakter plesiomorfik merupakan karakter primitive yang terdapat pada outgroup. Karakter sinapomorfik adalah karakter yang diturunkan dan terdapat pada kelompok monofiletik. Karakter morfologi telah lama digunakan dalam banyak penelitian filogenetika. Dengan pesatnya perkembangan teknik-teknik di dalam biologi molekuler penggunaan sekuen DNA dalam penelitian filogenetik telah meningkat pesat dan telah dilakukan pada semua tingkatan taksonomi, misalnya famili, marga, dan spesies. Filogenetik molekuler mengombinasikan teknik biologi molekuler dengan statistik untuk merekonstruksi hubungan filogenetika (Hillis et al., 1996).
Menurut Prakash (2007) Klasifikasi filogenetik dilakukan melalui konstruksi phylogeny tree. Phylogeny tree yang diperoleh merupakan hasil klasifikasi yang
menunjukkan hubungan filogenetik masing-masing strain bakteri yang diklasifikasikan. Terdapat 4 tahapan dalam mengkonstruksi phylogeny tree yang didasarkan atas data sekuens 16S rDNA, yaitu:
a. Preparasi sekuens 16S rDNA b. Aligment sekuens 16S rDNA c. Konstruksi phylogeny tree
d. Konstruksi matriks similaritas dan perbedaan nukleotida 16S rRNA.
B. Tujuan
Tujuan dari praktikum ini adalah mengetahui cara dan tahapan analisis kekerabatan bakteri dengan metode filogenetik molekuler.
II. MATERI DAN METODE A. Materi
Alat dan bahan yang digunakan dalam praktikum kali ini antara lain sekuesn nukleotida mikroba yang diujikan, dan komputer yang memiliki program PFE, MVSP, ClustalX, MEGA dan Ms.Words.
B. Metode
Metode yang digunakan dala praktikum taksonomi numerik-fenetik adalah sebagai berikut :
a. Koleksi data
1. Data sekuens 16S rRNA dari 15 isolat Flavobacetrium sp. dan satu isolat Pseudomonas sp. (outgroup) di download dari situs ncbi.
2. Masing-masing dicopykan ke program Words atau Notepad. Data tersebut berupa text file dan disimpan sebagai file.
b. Preparasi sekuens 16S rRNA
1. Data sekuens di atas selanjutnya dibuka dengan PFE (Programmer File Editor) dan sekuens dari masing-masing isolat diberi kode nama pada bagian depan sekuens (misal ‘flav 1, flav 2 dan seterusnya).
2. Selanjutnya di depan kode sekuens diberi tanda fasta format (>), hal ini agar dapat dibaca oleh program yang untuk melakukan alignment yaitu CLUSTALX.
3. Selanjutnya file disimpan dan diberi kode nama (save as), file ini sudah dalam bentuk fasta format file yang siap diload ke dalam CLUSTALX untuk dilakukan alignment.
c. Alignment sekuens 16S rDNA (ClustalX)
1. Data sekuens dari masing-masing strain (Cara kerja b) di load ke dalam program CLUSTALX untuk di align. Alignment bertujuan untuk menata sekuens agar satu sama lain diletakkan sesuai dengan posisi homologi antar sekuens. Artinya, daerah homolog harus diletakkan pada posisi yang sama (conserved region dengan conserved region, variable region dengan variable region). Dengan alignment antar sekuens gen 16S rRNA dari masing-masing strain dapat dibandingkan. Semua sekuens yang dialignment ditata dalam satu file oleh program ClustalX sebagai output file dalam beberapa pilihan format. Agar file hasil alignment dapat dibaca oleh program MEGA dan PHYLIP yang digunakan untuk mengkonstruksi phylogeny tree berdasarkan sekuens tersebut, maka file ini dibuat dalam phylip format (file
name.phy). Hal ini dapat dilakukan dengan memilih format output file pada waktu melakukan alignment dalam CLUSTALX.
2. Output file ini disimpan dalam direktori CLUSTALX (Misal C:\ClustalX\Flav.phy), sehingga setelah alignment dapat dicari hasil alignment berupa file yang diberi nama seperti contoh, dalam direktori ClustalX.
3. File ini selanjutnya digunakan untuk mengkonstruksi phylogeny tree dengan program MEGA atau PHYLIP.
d. Konstruksi Phylogeny Tree
Program MEGA 3.0.1
Seluruh sekuens 16S rDNA hasil alignment digunakan untuk mengkonstruksi phylogeny tree dengan program Mega 3.0.1.
1. Program Mega 3.0.1 dibuka, pilih alighment, lalu alighment explorer, kemudian pilih retrieve.
2. Selanjutnya mengambil file dengan format “.aln” hasil dari Clustalx. Klik file, lalu pilih Export alighment, pilih Mega format, dan save. Close, kemudian ketik 16S lalu “OK”, kemudian “no” dan pilih close. 3. Selanjutnya muncul perintah open data in mega? Pilih “yes”, lalu
minimize.
4. Pilih phylogeny, Construct phylogeny, pilih Neighbor Joining, lalu Test phylogeny, Bootstrap dengan replikasi 1000. Selanjutnya pilih compute.
II. HASIL DAN PEMBAHASAN
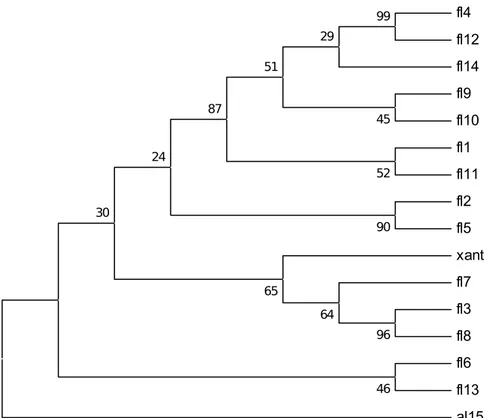
fl4 fl12 fl14 fl9 fl10 fl1 fl11 fl2 fl5 xant fl7 fl3 fl8 fl6 fl13 al15 96 99 64 65 90 46 30 24 52 87 45 51 29
Gambar 1. Hasil Dendogram Isolat Flavobacterium sp.
B. Pembahasan
Filogeni berasal dari bahasa Yunani yaitu Phylon yang artinya tribe atau clan dan Genesis yang berarti origin. Filogeni merupakan sejarah mengenai evolusi dari makhluk hidup yang digambarkan dalam bentuk pohon yang terdapat percabangan. Percabangan tersebut menggambarkan adanya divergensi. filogeni secara molekuler menggambarkan hubungan antar organisme berdasarkan susunan gennya dengan menggunakan sekuens DNA ataupun protein. Adanya ketidaksamaan dari sekuens genetik menggambarkan hasil divergensi atau adanya evolusi dari
organisme tersebut. Filogenik klasik hanya menggambarkan karakter morfologi dari suatu organisme. Analisis secara morfologi dapat dilihat dari sekuens DNA, sekuens RNA dan sekuens protein (Patwardhan, 2014).
Hal-hal yang harus diperhatikan dalam filogeni molekuler menurut Patwardhan (2014) diantaranya:
1. Kladogram merupakan pohon filogenetik yang menggambarkan adanya evolusi dari organisme.
2. Hompolasy merupakan sekuens yang sama akibat hasil konvergensi tetapi tidak menggambarkan adanya evolusi secara langsung.
3. Internal Transcribed Spacers (ITS) yaitu rRNA akan ditranskripsi menjadi single transcript oleh ITS ini.
4. Monofiletik yaitu taxa dalam pohon filogenetik mempunyai satu nenek moyang yang sama.
5. Outgroup suatu taxa yang menjadi karakter pembanding dalam pembuatan pohon filogenik.
Basic Local Alignment Search Tool (BLAST) dari NCBI ialah program yang berfungsi mencari program database urutan biologis. Program ini menggunakan algoritma untuk membandingkan antara DNA sekuens protein untuk query database. BLAST merupakan perangkat bioinformatika yang berkaitan erat dengan penggunaan pangkalan data sekuens biologi. Penelusuran BLAST (BLAST search) pada pangkalan data sekuens memungkinkan ilmuwan untuk mencari sekuens baik asam nukleat maupun protein yang mirip dengan sekuens tertentu yang dimilikinya. Hal ini berguna untuk menemukan gen sejenis pada beberapa organisme, memeriksa keabsahan hasil sekuensing, atau untuk memeriksa fungsi gen hasil sekuensing. Algoritma yang mendasari kerja BLAST adalah penyejajaran sekuens (Rizki, 2008).
Proses analisis filogenik molekuler dibutuhkan beberapa software, diantaranya:
1. Programmer File Editor (PFE) (Felsenstein, 1993)
Merupakan program yang berguna untuk menata data sekuen DNA sehingga sesuai dengan bentuk data yang dapat dibuka pada program ClustalX.
2. MEGA / Phylip (Felsenstein, 1993)
Merupakan program yang digunakan untuk mengkonstruksi Phylogeny tree sesuai dengan algoritma evolusi sehingga dapat divisualisasikan dengan jelas. 3. ClustalX (Felsentein, 1993)
Merupakan program yang diperlukan untuk menata (alignment) data sekuen dari berbagai strain organisme sehingga dihasilkan pasangan sekuen antar strain dimana daerah conserved akan mengumpul dan daerah variabel juga saling mengumpul. Output dari program ini berupa file dalam format .*aln, .*GDE dan *.phy.
4. Phydit (Chun, 1999)
Merupakan program yang digunakan untuk mengolah data sekuen yang kemudian akan di align untuk mendapatkan data berupa matriks similaritas nukleotida dan perbedaan nukleotida. Matriks yang dihasilkan program Phydit dapat dilihat di program NotePad dan dipindah dalam program Microsoft Excel.
Tahapan dalam analisis karakter mikroba menggunakan filogenik molekuler yang pertama adalah pengkoleksian data dengan mencari sekuens 16S rRNA dari 15 isolat yang sudah ditentukan, misalnya Flaboctarium sp. dan Pseudomonas sp. sebagai outgroup yang di download dari situs ncbi kemudian dicopykan ke dalam notepad. Langkah selanjutnya melakukan preparasi sekuens 16S rRNA dengan menggunakan PFE (Programmer File Editor) dan sekuens dari masing-masing isolat. Selanjutnya, tahap alignment sekuens 16S rRNA menggunakan software CLUSTALX. Alignment bertujuan untuk menata sekuens agar satu sama lain diletakkan sesuai dengan posisi homologi atau sekuens. File disimpan dalam format .phy agar dapat dibaca oleh program MEGA. Langkah selanjutnya adalah konstruksi Phylogeny Tree menggunakan program MEGA dengan memilih Construct phylogeny, kemudian Neighbor Joining, lalu Test phylogeny, Bootstrap dengan replikasi 1000. Selanjutnya pilih compute. Langkah terakhir adalah konstruksi matriks similaritas dan perbedaan nukleotida 16S rRNA menggunakan aplikasi PHYDIT (Rizki, 2008).
Isolat yang dipakai dalam praktikum kali ini adalah 15 isolat Flavobacterium sp. berbagai strain dengan outgroup berupa Pseudomonas sp. didapat hasil bahwa strain fl4 dan fl12 mempunyai nilai similaritas yang tinggi dengan nilai 99% artinya kedua strain ini mempunyai kekerabatan yang dekat. Strain yang memiliki kekerabatan yang dekat terlihat pada fl2 dengan fl5 dengan indeks kesamaan 90%. Strain fl3 dan fl8 mempunyai nilai kesamaan 96%. Strain fl6 dan strain fl13 mempunyai indeks similarias sebesar 46% artinya kedua strain tersebut berbagi karakter sehingga nilai kesamaan tidak terlalu tinggi. Strain fl1 dan fl11 mempunyai indeks kesamaan sebesar 52%. Indeks similaritas paling kecil terlihat pada strain fl9
dan fl10. Flavobacterium sp. mempunyai karakter berbentuk basil, Gram negatif, fakultatif anaerob. Saat kondisi aerob bakteri ini mampu mengoksidasi asam amino, sedangkan pada kondisi anaerob bakteri ini dapat melakukan fermentasi. Flavobacterium sp. mempunyai range suhu dan pH yang luas. Suhu flavobacterium sp. dapat tumbuh adalah 10-400C (Bergey’s Manual, 2010).
IV. KESIMPULAN DAN SARAN A. Kesimpulan
Berdasarkan hasil dan pembahasan maka dapat disimpulkan bahwa :
1. Langkah yang digunakan dalam analisis filogenetik molekuler yaitu koleksi data dari ncbi, preparasi sekuens 16S rRNA menggunakan PFE, alignment sekuens 16S rRNA menggunakan ClustalX, konstruksi phylogeny tree menggunakan MEGA 3.0.1 dan langkah terakhir adalah konstruksi matriks similaritas menggunakan PHYDIT.
2. Hasil dari analisi molekuler dengan isolat Flavobacetrium sp. berbagai strain didapat data strain fl4 dan fl12 mempunyai nilai similaritas yang paling tinggi 99%, dan indeks similaritas paling kecil terlihat pada strain fl9 dan fl10 dengan nilai similaritas 45%.
B. Saran
Jurnal yang dipakai untuk praktikumnya mungkin lebih baik jika 1 kelompok 1 jurnal tidak 1 orang 1 jurnal agar lebih efisien dalam pengerjaannya tidak memakan waktu yang lama. Terima kasih sudah membagi ilmunya, semoga lebih baik lagi di praktikum selanjutnya.
DAFTAR REFERENSI
Chun, J.. 1999. Phylogenetic Editor (PHYDIT) Windows Version. New York Press, New York.
Felsenstein, J.. 1993. Phylogeny Inference Package version 3.5 c.London.
Hillis, D.M., C. Moritz, and B.K. Mable. 1996. Molecular Systematic. 2nd Ed. Massachusetts: Sinauer Assocites.
Patwardhan et al. 2014. Phylogenetics & Evolutionary Biology Molecular Markers in Phylogenetic Studies A Review. J., Phylogen Evolution Biol, 2 (2) : 2-9. Prakash, O., Verma, M., Sharma, P., Kumar, M., Kumari, K., Singh, A., Kumari, H.,
Jit, S., Gupta, S.K., Khanna, M. and Lal, R. 2007. Polyphasic approach of bacterial taxonomy. Second edition. Chapman & Hall, London.
Rizki, S. 2008. Bioteknologi Tanaman. Universitas Brawijaya Press, Malang.
Schleifer, K.H. 2009. Classification of Bacteria and Archaea: past, present and future. Systematic and Applied Microbiology 32: 533-542.
Sembiring, L.. 2010. Sistematika molekular (BIO 765), Petunjuk Praktikum. Laboratorium Mikrobiologi Fakultas Biologi UGM, Yogyakarta.
Woese, C.R. 1987. Bacterial evolution. Microbiol Rev. 51: 221-272.
KLASIFIKASI MIKROBA (Flavobacterium sp.) DENGAN METODE FILOGENETIK MOLEKULER
Oleh :
Nama : Silviyatun Ni’mah
NIM : B1J013016
Kelompok : 1 Rombongan : I
Asisten : Hedi Susanto
LAPORAN PRAKTIKUM SISTEMATIKA MIKROBA
KEMENTERIAN RISET TEKNOLOGI DAN PENDIDIKAN TINGGI UNIVERSITAS JENDERAL SOEDIRMAN
FAKULTAS BIOLOGI PURWOKERTO