20
NEUROFIBROMATOSIS
Rr. Suzy Indharty
Pendahuluan
Neurofibromatosis yang juga dikenal sebagai von recklinghausen disease, merupakan suatu kelainan genetic yang memberi efek pada berbagai organ tubuh, terutama kulit dan sistem saraf. Beberapa terjadi saat lahir, tetapi yang lain terjadi setelah dewasa. Terdapat tiga bentuk neurofibromatosis, yaitu NF1, NF2, dan Schwannomatosis.
Neurofibromatosis (NF) 1 atau
yang dikenal sebagai peripheral
neurofibromatosis merupakan jenis yang paling sering ditemukan, yaitu sekitar 1/3000-4000 orang. NF1 memiliki ciri khas berupa banyak bercak lahir, dan tumor pada sistem saraf dan otak. NF1 merupakan suatu penyakit autosomal dominant yang diturunkan. Namun ditemukan 50% penderita yang tidak berhubungan dengan turunan keluarga dan merupakan hasil dari suatu mutasi gen, yaitu suatu perubahan baru yang tidak ditemukan pada anggota keluarga yang lain.
Neurofibromatosis (NF) 2 yang
diketahui sebagai central
neurofibromatosis atau bilateral acoustic neurofibromatosis ditemukan sekitar 10% dari seluruh penderita NF dengan insiden
1/150.000 jiwa. Tumor pada saraf pendengaran yang biasanya mengenai kedua telinga (auditory nerve). Penderita NF2 dapat juga memunyai lesi yang lain, seperti tumor otak dan spinal cord. Mayoritas penderita NF2 hasil dari mutasi gen dengan perubahan yang baru, dan tidak ditemukan pada anggota keluarga yang lain. Schwannomatosis adalah suatu bentuk lain dari NF yang jarang. Jenis ini baru dikenal dan tidak seperti NF1 dan NF2.
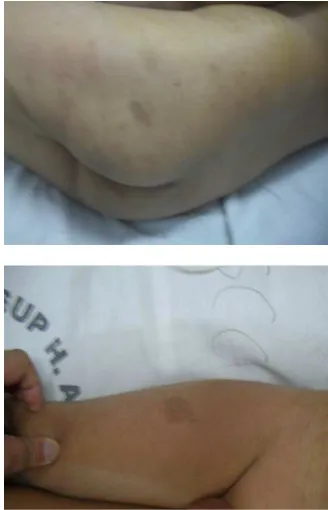
Neurofibromatosis tipe I memiliki ciri khas berupa café-au-lait spots, perkembangan bagian mata yang disebut iris Lisch nodules, lesi rubbery pada kulit yang disebut neurofibroma. Lesi ini dapat terjadi juga pada dermatofibromas, multiple optic nerve gliomas, bilateral plexiform neurofibroma, dan malignant peripheral nerve sheath tumor.
Epidemiologis
NF 1 merupakan salah satu kelainan genetic yang terbanyak, dengan insiden 1 dari 3000-4000 orang.1 NF 1 atau von Recklinghausen’s disease merupakan
suatu penyakit autosomal dominant
Sekitar 50% dari kasus NF1 timbul secara sporadic karena terjadi mutasi baru. NF1 merupakan satu dari kebanyakan kelainan single gene. Kelainan ini memunyai turunan fenotipe yang tinggi, sehingga orang tua yang tidak memberikan efek, memunyai risiko rekuren yang rendah. Kebanyakan NF1 dapat dideteksi pada bayi dengan berdasarkan pada suatu kelainan kulit yang biasanya makin jelas
dengan pertambahan usia, terutama
setelah pubertas. Hampir 100% NF1 menunjukan penetrasi pada usia delapan tahun.3 Sindroma ini disebabkan oleh mutasi gen dari kromosom 17q11.2 dengan kode protein besar disebut neurofibromin. Bagian dari protein ini, yaitu GTPase-activator yang berperan sebagai signal transduction melalui perubahan yang menguntungkan dari bentuk aktif GTP-bound dari ras dan
menghubungkan G-protein kebentuk
inaktif GDP-bound.4,5 Fungsi gen NF1
sebagai gen supresi tumor dalam
inaktivasi ke dua allele diperlukan untuk tumorigenesis. Penderita dengan NF1 lahir dengan hanya satu kopi normal dari gen dan, yang lain mutasi atau hilang menyebabkan inaktif allele ke dua dan secara teori cukup untuk pembentukan tumor. NF2 suatu turunan pada autosomal dominant dengan insiden 1:37.000, dan
tanpa adanya predileksi kelamin.
Umumnya penderita NF2 memberikan gejala pada usia pubertas, namun onset usianya sangat bervariasi. Onset gejala pada usia menengah sekitar tujuh belas tahun biasanya dengan tinnitus dan/atau hilangnya pendengaran akut akibat tumor vestibular.
Diagnosis
Sebagian besar diagnosis NF1
berdasarkan pemeriksaan klinis yang memperlihatkan gambaran berupa café-au-lait spot, Lisch nodules (pigmentasi
iris hamartoma), axillary dan inguinal freckling, skeletal lesions, seperti sphenoid wing dysplasia dan penipisan cortex tulang panjang, dan optic glioma,
serta meningginya insiden Central
Nervous System (CNS), dan tumor sistemik lainnya.6 Seringnya perubahan warna kulit yang timbul terjadi sebelum remaja (95% dari penderita dengan NF1). Dermal neurofibroma timbul dari sel schwann dan terjadi sekitar 99% dari penderita. Tumor ini timbul selama remaja dengan pertambahan jumlah dan ukurannya sesuai usia. Beberapa gejala
yang lain termasuk macrocephaly,
perubahan pada pembuluh darah, tubuh
pendek, scoliosis, dan gangguan
pendengaran.
Diagnosis NF1 jika ditemukan dua atau lebih kriteria ini, yaitu:
1. Enam atau lebih café au lait spot (diameter lebih dari lima mm sebelum pubertas dan lebih dari lima belas mm setelah pubertas). 2. Dua atau lebih neurofibroma dari
segala tipe dengan satu atau lebih plexiform neurofibroma
3. Freckling pada axilla atau daerah inguinal (tanda Crowe’s)
4. Dua atau lebih Lisch nodule (iris hamartoma)
5. Suatu tumor jaras optik
6. Lesi tulang, seperti sphenoid wing dysplasia, atau penipisan cortex tulang panjang, dengan atau tanpa pseudoarthrosis
7. Keturunan tingkatan pertama
(orang tua, saudara kandung, atau anak cucu) dengan NF1 melalui kriteria tersebut di atas
BAB 20 NEUROFIBROMATOSIS
285 NF1 memunyai tanda glioma pada jaras
optic.7 Hal ini memberi kesan bahwa banyak penderita NF1 yang tanpa gejala.
Tumor dengan gejala umumnya
menunjukan berkurangnya visual acuity, defek lapangan pandang, proptosis, dan precox puberty akibat kompresi hypothalamus. NF1 berhubungan dengan
bertambahnya insiden glioma
parenchyma terutama pada brain stem, cerebellar peduncle, globus pallidus, dan midbrain. Sifat biologis glioma brain stem pada penderita NF1 berbeda secara signifikan dengan penderita tanpa NF1. Umumnya penderita glioma brain stem dengan NF1 memunyai hasil akhir yang lebih baik dibandingkan dengan anak yang tanpa NF1. Tumor brain stem jangan dibingungkan dengan perubahan white matter yang tidak spesifik karena sering ditemukan pada MRI penderita dengan NF1 dan klinis yang tidak diketahui.8
Penderita NF1 timbul tidak hanya pada CNS, tetapi juga pada sistem saraf
perifer. Neurofibromatosis dan
schwannoma lebih sering timbul dari saraf perifer utama, seperti saraf ulna dan radius. Transformasi tumor malignant cenderung pada saraf perifer. Tumor ini terjadi kurang dari 5% pada anak-anak dengan NF1, tetapi cenderung memiliki mortalitas yang tinggi pada orang dewasa dengan NF1.9 Sheath tumor saraf perifer yang ganas merupakan sarcoma yang bersifat kemoresisten dan berhubungan dengan angka survival lima tahun yang rendah walaupun telah diterapi secara agresif. Tanda klinisnya berhubungan
dengan transformasi ganas dan
pertumbuhan yang cepat. Indikasi operasi pada tumor yang besar adalah masalah kosmetik. Penderita dengan NF1 juga
memunyai risiko tumor non-CNS,
termasuk Wilm’s tumor,
rhabdomyosarcoma, leucemia,
melanoma, medullary thyroid carcinoma, dan pheochromacytoma.
Anak dengan NF2 selalu
diagnosis saat dewasa muda dengan keluhan hilangnya pendengaran atau telinga berdengung. Bilateral vestibular schwannoma merupakan lesi khas pada penderita NF2, biasanya dengan tinnitus atau hilangnya pendengaran.10 Tumor ini ditemukan sekitar 96% pada penderita NF2 dengan 90% bersifat bilateral, dan 6% bersifat unilateral. Vestibular Schwannoma yang disebut juga accoustic neuroma memiliki sebutan yang tidak tepat karena tumor timbul dari sel-sel Schwann dan khas melibatkan vestibular dibandingkan dengan cabang accoustic (cochlear) dari saraf kranial ke delapan.
Penderita NF2 secara keseluruhan
menunjukan predileksi tumor meninges dan sel Schwann. NF2 dapat melibatkan saraf facial, saraf trigeminal, dan berbagai
saraf spinal, seperti Schwannoma,
meningioma, dan retinal hamartoma. Gejala yang sering ditemukan termasuk
hilang pendengaran, tinnitus, dan
gangguan keseimbangan dari vestibular Schwannoma. Usia saat timbulnya gejala, yaitu antara lima belas sampai tujuh puluh empat tahun. Penderita NF2 di bawah usia sepuluh tahun sering disertai dengan defisit penglihatan atau pertumbuhan tumor kulit. Penderita NF2 juga dapat
mengalami pertumbuhan tumor
paraspinal yang menekan spinal cord dan disertai dengan myelopathy. Lesi ini sering ditemukan pada penderita NF2 dan
merupakan sumber morbiditas dan
mortalitas.11 Lesi yang berhubungan
dengan NF2 termasuk posterior
subcapsular cataract (63%), retinal hamartoma, optic nerve-sheath meningioma, meningioma, ependymoma (biasanya spinal cord), glioma, dan trigeminal schwannoma.
50.000 orang.12 Dasar petunjuk perubahan genetic pada kelainan ini diketahui
melalui studi sporadic vestibular
neurilemoma dan meningioma, yaitu tumor yang khas dari NF2. Penelitian
tersebut menunjukan bahwa sering
ditemukan hilangnya lengan panjang dari kromosom 22. Observasi cytogenic ini telah dilakukan dan mengidentifikasi adanya daerah mutasi pada 22q12.13 Produksi dari gen ini adalah protein merlin (moesin, ezrin, dan radixin-like protein) atau schwannomin, yaitu suatu polipeptida yang terlibat dalam hubungan elemen sitoskeletal dengan membran plasma protein.14 Teknik molecular mampu mengidentifikasi mutasi NF1 lebih dari 95% kasus. Ini dilakukan apabila secara klinis tidak dapat ditegakan diagnosis secara pasti. Diagnosis prenatal dapat juga ditemukan dengan adanya riwayat keluarga yang positif NF1.
Kriteria diagnosis berdasarkan penggunaan fasilitas untuk NF2 berbeda
dengan NF1 secara keseluruhan.
Diagnosis secara laboratorium harus ditemukan adanya mutasi DNA pada gen NF2, dan menunjukan adanya hubungan dengan DNA asal setidaknya dari dua anggota keluarga. Penderita memunyai kombinasi antara saraf kranial VIII dan saraf lainnya, seperti neurilemoma, meningioma, glial neoplasma, neurofibroma, dan juvenile posterior subcapsular cataract. Walaupun café-au-lait macules dan cutaneous neurofibroma kadang-kadang ditemukan, tetapi lesi ini hanya sedikit dibandingkan dengan pada penderita dengan NF1, dan bukan termasuk kriteria dalam diagnosis.
Sifat-sifat NF1 dan NF2
memperlihatkan tanda yang terbatas terhadap beberapa segment tubuh. Pada kebanyakan situasi, penderita memunyai café-au-lait macules dan neurofibroma pada satu ekstremitas atau setengah badan, dan memunyai Lisch nodules yang
ipsilateral pada mata. Kelainan ini disebut
bentuk segmental dari NF1 yang
ditemukan sekitar 5% dari penderita NF115. Hal ini timbul secara mosaicism, yaitu mutasi gen NF1 yang terjadi beberapa waktu setelah fertilisasi saat perkembangan embryo. Bentuk segmental NF2 juga tampak pada penderita dengan
berbagai ciri neurilemoma yang
melibatkan saraf perifer dari ekstremitas tanpa gambaran sentral dari NF2.16
Manifestasi Non-Neurological
Karena kedua NF1 dan NF2 merupakan kelainan multisystem maka penderita selalu datang berobat dengan gejala dan tanda yang tidak langsung menunjukan suatu tumor sistem saraf.
BAB 20 NEUROFIBROMATOSIS
287
NF1
Café-au-lait macule dan axillary freckling selalu menjadi sumber pertimbangan karena klinisnya tidak serius. Lesi ini dihasilkan dari dari koleksi pigmen melanin terutama di abdominal, tetapi
tanpa disertai komponen tumor.
Melanocytic iris hamartomas menyerupai lesi Lisch nodule. Lesi ini bertambah selama remaja tanpa disertai penglihatan yang terganggu namun lebih dari 90% menghilang setelah pubertas.
Displasia kongenital dari tulang panjang terutama tibia cenderung menyebabkan
fraktur patologis yang sulit
penyembuhannya. Pseudoartrosis sangat sulit untuk dikoreksi dan beberapa
penderita memerlukan amputasi.17
Osseous dysplasia dapat juga melibatkan tulang sphenoid sebagai proses kongenital
atau didapat yang cenderung
menyebabkan herniasi lobus temporalis ke orbita dan pada beberapa kasus
menimbulkan pulsasi proptosis dan
kejang. Karena beberapa penderita
memperlihatkan kerusakan progresif
terhadap deformitas ini, maka intervensi operasi dilakukan terbatas pada anak-anak dengan proptosis yang berat dan disertai orbital plexiform neurofibroma atau
intractable seizures akibat keterlibatan lobus temporalis. Pada kasus yang jarang, rekonstruksi menggunakan split-thickness calvarial grafts atau rib grafts bisa bermanfaat.
Manifestasi pada spinal juga sering, walaupun tidak ditemukan keterlibatan neoplasma. Beberapa tingkatan scoliosis juga ditemukan terutama pada penderita dengan NF1, namun tidak semua kasus memerlukan terapi khusus.16 Sebagian kecil anak-anak NF1 disertai dengan scoliosis yang berat dan cepat progresif.18 Karena banyak penderita ini memunyai intra-axial atau extra-axial neurofibroma, maka perlu ditekankan hubungannya dengan prosedur spine-straightening dengan MR-scan sebagai dasar evaluasi sebelum operasi. Manifestasi spinal lain yang kurang serius termasuk vertebral scalloping dan nonneoplastic widening dari neural foramina pada dural ectasia
dan pembentukan meningocele.
Umumnya tidak diperlukan intervensi khusus setelah sangkaan neoplasma telah disingkirkan melalui pemeriksaan MR-scan atau CT-myelography.
Sebagai tambahan bahwa
penderita NF1 dengan segmental
hipertropi dapat melibatkan salah satu bagian tubuh, seperti kepala atau satu
ekstremitas.19 Walaupun biasanya
komponen tumor yang mendasari daerah yang terlibat, deformitas selalu melebihi dan langsung melibatkan tumor. Ini masih belum pasti apakah gambaran umum dysplasia mesenchymal terlibat dalam area ini atau merupakan awal kombinasi faktor neurogenic dan humoral oleh tumor.
Penderita dengan NF1 juga
memunyai risiko terjadinya malignansi pada berbagai sistem. Studi terbaru menunjukkan risiko relatif chronic myelomonocytic leucemia diantara penderita NF1 sangat mengejutkan, yaitu 221 kali dibandingkan dengan populasi umum.20 Tumor lain yang meningkat frekuensinya pada penderita NF1, yaitu neurofibrosarcoma, pheochromocytoma, rhabdomyosarcoma, adeno-carcinoma dari ampulla Vater, melanoma, non
Hodgkin’s lymphoma, dan lymphoblastic leucemia.19,21
NF2
Manifestasi nonneural yang paling serius dari NF2 adalah posterior subcapsular cataracts. Kelainan ini ditemukan sebesar 85% dari penderita NF2 dan selalu bertambah dengan usia.22 Karena lesi ini dapat mengancam penglihatan shingga
diperlukan pemeriksaan mata dan
mungkin operasi katarak bila indikasi.
Penderita dengan hilangnya satu
penglihatan akibat lesi ini, maka diperlukan perhatian dan pengawasan untuk mencegah kerusakan penglihatan mata yang satu lagi. Pengwasan juga dilakukan pada fungsi saraf wajah untuk
mempertahankan mata tertutup dan
mencegah kerusakan cornea.
signifikan. Penderita dengan NF1
memperlihatkan berbagai manifestasi nonneoplatic neurological yang harus dibedakan dari tumor-tumor lain untuk
menghindari intervensi yang tidak
diperlukan.
NF1
Fokal Area Dari Meningginya T2 Signal
BAB 20 NEUROFIBROMATOSIS
289 keduanya, dan paling sering tampak di
basal ganglia, internal capsule, brainstem, dan cerebellum.21 Banyaknya
foci ini dapat digunakan sebagai
tambahan kriteria diagnosis. Karena konfirmasi biopsi tidak lazim dilakukan maka penjelasan spekulatif ini disebut unidentified bright objects (UBO) termasuk foci dari low-grade glioma, hamartoma, heterotopias, dan abnormal myelination.23 Dilaporkan bahwa foci dari T2 signal abnormalities selalu bertambah frekuensi dan jumlahnya pada masa kanak-kanak dan berkurang menjelang dewasa muda. Lesi ini menggambarkan bahwa usia berhubungan dengan kelainan dalam mielinisasi. Ada yang mendapatkan
bahwa kelainan signal berhubungan
dengan deteksi learning disabilities. Hal ditemukan setidaknya 25% dari penderita NF1, namun tidak ditemukan pada studi lain.24
Lesi Jaras Optik
Kedua tersering kelainan foto radiologis
pada penderita NF1 adalah optic
hypothalamic glioma dan ditemukan sebesar 15%. Beberapa tipe lesi dapat di-jumpai, baik satu tipe atau dalam bentuk kombinasi. Kelainan yang paling ringan berupa penebalan satu atau kedua saraf optik, walaupun kebanyakan lesi adalah low-grade glioma. Kelainan lain yang juga bisa ditemukan adalah hyperplasia dari optic nerve sheath. Pada penderita lain diperoleh penebalan globular saraf optik dan chiasma yang berhubungan dengan T2 signal abnormalities yang melintas ke belakang sepanjang jaras optic dan ke atas arah hypothalamus. Pada biopsi ditegaskan adanya low-grade glioma.25 Sebagian kecil penderita
memperlihatkan massa besar yang
melibatkan optic chiasma dan
hypothalamus yang meluas ke atas arah ventrikel III, ke samping arah fossa
temporalis, ke depan di bawah lobus frontalis, dan ke belakang daerah perimesencephalic.
Manajemen yang optimal
terhadap lesi ini masih kontroversi. Hanya 10-20% dari lesi ini yang memperlihatkan pembesaran ukuran tumor atau klinisnya memburuk. Kebanyakan penderita tidak memunyai gejala dan memperlihatkan sedikit frekuensi pembesaran tumor, sehingga foto rutin masih menjadi masalah. Ada beberapa pendapat ahli yang menganjurkan dilakukan MR-scan setiap tahun pada anak dengan NF1, sedangkan pendapat yang lain masih menganjurkan foto pada penderita apabila ada gejala dan tanda yang progresif,
seperti asimetris atau monocular
nystagmus, strabismus, visual loss, visual field deficit, precocious puberty, growth delay, diecephalic syndrome, sakit kepala, dan peningkatan TIK, tetapi untuk evaluasi klinis dan pemeriksaan neuro-ophthalmology perlu dilakukan setiap tahun.
Dengan bertambahnya
pengetahuan mengenai optic pathway lesions pada penderita NF1, maka indikasi intervensi operasi sangat kecil. Gambaran histologis dari lesi ini jarang berbeda, oleh karena itu maka biopsi untuk diagnosis tidak diperlukan. Penderita dengan unilateral optic nerve glioma, proptosis, dan hilangnya penglihatan merupakan indikasi operasi reseksi saraf yang terlibat pada bola mata hingga chiasma. Kasus yang sangat jarang terjadi karena pada MR scan ditemukan juga keterlibatan chiasma dan optik nerve kontralateral. Dalam hal ini, radiotherapy dan kemoterapi kemungkinan lebih baik
dilakukan. Harus dibedakan juga
pada anak-anak dengan pertumbuhan tumor exophytically lebih besar dari chiasma opticum.25 Masih belum dapat dipastikan apakah hasil jangka panjang tercapai dengan reseksi agresif bila dengan tanpa operasi.26
Penatalaksanaan intervensi
operasi masih berperan walaupun terdapat
kontroversi. Chiasmal hypothalamic
glioma jelas bukan merupakan kandidat
untuk dilakukan operasi karena
keterlibatan optic dan hypothalamus yang difus. Tindakan radiotherapy dilakukan pada lesi yang tidak berindikasi operasi.27 Cara ini memberikan hasil yang baik, karena lesi stabil, dan kadang-kadang lesi mengalami regresi dengan perbaikan fungsi penglihatan yang signifikan.
Radiasi dapat juga menyebabkan
gangguan endokrin dan congnitive yang berat serta risiko mengalami radiation-induced malignancies dan vasculopathy seperti sindroma moyamoya.28
Berbagai regimen telah digunakan dengan respon rata-rata 20-80% dan stabilization rata-rata 75-100%.27 Studi lain dengan pemberian casboplatin-vincristine dan kombinasi 6-thioguanine, procarbazine, dibromodulcitol, cyclohexylnitrosourea (CCNU), dan vincristine memberikan hasil yang lebih baik. Beberapa studi lain menemukan
bahwa respon kemoterapi tidak
memberikan perbedaan signifikan pada anak-anak dengan NF1 dan tanpa NF1.29
Cerebral Dan Cerebellar
Hemispheric Gliomas
Sedikit sekali penderita dengan NF1 lesinya membesar ke dalam hemisphere cerebral dan cerebellar. Lesi itu seperti terutama glioma yang memperlihatkan efek massa lokal, dan berkurangnya sinyal
pada T1-weighted images dengan
enhancement yang uniform, ring-like
sebagai mural nodule, atau tidak
ditemukan semuanya. Dibandingkan
dengan chiasmal-hypothalamic gliomas, lesi ini jarang ditemukan pada penderita NF1 sehingga jarang dibicarakan. Lebih dari setengah tumor non-optic histologis
memberi gambaran malignant dengan
persentase yang tinggi pada anak-anak tanpa NF1 secara signifikan. Sebaliknya
studi lain mendapatkan lesi yang
mayoritas jinak, dan dapat dilakukan reseksi total atau hampir total. Oleh sebab itu lebih baik dilakukan tindakan reseksi total yang agresif pada hemisphere cerebral dan cerebellar. Berbagai modalitas tambahan saat intraoperasi, seperti stereotactic, USG, dan fungsional monitoring dapat memberi hasil yang lebih baik.
Penatalaksanaan setelah operasi
bergantung pada hasil pemeriksaan
histologis. Penderita dengan tidak dapat direseksi atau rekuren setelah reseksi agresif, diberi tambahan terapi dengan radiotherapy atau kemoterapi. Penderita NF1 yang berkembang menjadi malignant glioma ditangani sama seperti penderita tanpa NF1. Setelah dicoba reseksi secara maksimal, kemudian dilakukan kombinasi
terapi dengan radiotherapy dan
kemoterapi pada penderita dengan usia di atas tiga tahun. Awalnya penderita diberi
kemoterapi dengan harapan radiasi
ditunda selama mungkin. Meskipun jelas kemoterapi sangat bermanfaat untuk malignant glioma terutama glioblastoma multiforme, namun dosis maksimal terapi kombinasi masih belum bisa dipastikan.
Brain Stem Gliomas
Penderita brainstem glioma dengan NF1
adalah kelompok heterogen yang
BAB 20 NEUROFIBROMATOSIS
291 dengan meningkatnya signal T2-weighted
images.30
Kelompok lain dari lesi batang otak pada penderita dengan NF1 adalah enhance nodule fokal dengan atau tanpa
hubungan dengan daerah kista.31
Meskipun diperkiraan lesi low-grade pilocytic astrocytoma berdasarkan scan,
namun konfirmasi histologis tetap
diperlukan. Sifat biologis dari lesi adalah ini secara umum juga pertumbuhannya lambat tetapi tidak dapat diramalkan pada akhirnya. Dilakukan terapi pada anak-anak dengan tumor yang membesar secara progresif dan yang memiliki efek lokal
yang signifikan atau dengan
perkembangan gejala klinis yang
progresif. Meskipun ditemukan massa yang kecil, lesi fokal intrinsic tersebut membesar secara progresif, dan tidak ada gejala selama remaja yang kemudian regresi spontan tanpa pengobatan. Karena riwayat alami dari beberapa lesi masih belum diketahui, maka dilakukan scan rutin pada penderita dengan terapi konservatif.
Kelompok lesi batang otak pada penderita NF1 adalah periaquaductal glioma. Lesi ini bermanifestasi dengan aquaductus stenosis dan diperkirakan low-grade glioma atau glial hamartoma, walaupun konfirmasi biopsi terbatas.32 Pertumbuhan tumor ini lambat dan secara umum tetap tumbuh selama beberapa paraspinal dan peripheral neurofibroma. Tumor ini dahulu dikategorikan kedalam neurilemoma yang merupakan tanda dari
NF2. Kedua kelompok tumor ini
dibedakan berdasarkan atas analisis histologis.33 Karakteristik neurilemoma
memperlihatkan adanya daerah dari sel (Antoni A) dengan arsitektur palisading spindle cells yang orientasi ke verocay bodies, dan Antoni B mengatakan adanya loose array of spindle cells yang
dilatarbelakangi oleh mucinous.
Sebaliknya, neurofibroma terdiri atas spindle cells dalam myxomatous stroma dan bergabung dengan axon myelinated dan unmyelinated yang jarang ditemukan pada neurolemoma.
Lesi ini dapat dilihat dengan
menggunakan MR scan untuk
menggambarkan hubungan diantara
tumor, sekitar saraf, dan struktur-struktur yang berdekatan.34 Pada penelitian ini, penderita dengan lesi paraspinal memberi informasi tentang luasnya gangguan foraminal dan intraspinal, dan untuk penderita dengan lesi visceral memberi informasi tentang hubungan antara tumor dan struktur sekitar yang penting. Berdasarkan gambaran scan, lesi ini bisa
dikategorikan melalui susunan
pertumbuhannya sebagai fusiform
neurofibroma yang lesinya berlainan dan melibatkan daerah sekitar saraf, serta plexiform neurofibroma memperlihatkan keterlibatan yang meluas ke satu atau dua saraf.34
pembesaran yang cepat. Dapat juga timbul di daerah punggung yang bisa menyebabkan iritasi. Hanya sedikit dari tumor ini yang akan menjadi ganas (seperti neurogenic sarcoma), tetapi penderita harus melaporkan lesi yang cepat membesar, warna kemerahan atau ulkus, dan menggangu secara progresif setelah eksisi biopsi.
Peripheral nerve neurofibroma menimbulkan gejala disfungsi neurologis pada saraf utama dengan rasa sakit dan parasthesia. Hal ini selalu merupakan awal atau eksaserbasi melalui manipulasi atau perkusi saraf yang terlibat.34 Tidak
seperti neurilemoma yang khas
melibatkan satu dari single fascicle saraf utama dan fascicle lain pada kapsul tumor, neurofibroma selalu melibatkan banyak atau semua fascicle saraf, dan pada berbagai kasus terbatas reseksinya. Penanganan lesi ini masih kontroversial dan sebagian ahli merekomendasi biopsi dan observasi, sedangkan ahli yang lain menganjurkan blok reseksi dengan end to end atau graft terhadap saraf yang terlibat, atau intracapsular enucleation.35 Banyak kasus memungkinkan untuk dilakukan reseksi multiple yang melibatkan fascicle tanpa mengorbankan fungsi saraf.34 Tindakan ini dilakukan diseksi pada bagian proximal dan distal dari tumor dan identifikasi fascicle asal timbulnya tumor kemudian dibuang. Penggunaan teknik rangsangan saraf saat operasi untuk konfirmasi fascicle dan pengangkatan tumor sesungguhnya tidak berguna. Pada beberapa kasus, reseksi total tidak
mungkin tanpa risiko kerusakan
neurologis, dan reseksi sebagian harus mengikuti arah pertumbuhan massanya. Pada keadaan seperti ini, perkembangan sisa tumor harus dijaga lebih lanjut karena sekitar 15% tumor mengalami perubahan menjadi ganas.34 Penanganan operasi dan setelah operasi menjadi ganas ini harus melibatkan beberapa disiplin ilmu, seperti
neurologis, general surgery, orthopedic, medical oncologic, dan radiation oncologic.36
Reseksi total tidak mungkin pada tumor plexiform karena infiltrasi yang luas sepanjang saraf yang terlibat. Jika hanya satu saraf yang terlibat, reseksi en bloc dan graft secara teori memungkinkan tetapi hasil fungsionalnya buruk. Ini bukan merupakan pilihan untuk plexus neurofibroma yang melibatkan plexus brachial dan lumbosacral. Reseksi biasanya sebagai pilihan terakhir untuk lesi yang ganas dan menyakitkan atau adanya kompresi struktur sekitarnya yang menimbulkan gejala berat. Karena lesi dapat memperlihatkan sifat yang lambat maka terapi agresif hanya terbatas untuk tumor yang menyebabkan kerusakan progresif.
Paraspinal neurofibroma biasanya berbentuk tumor fusiform yang melibatkan cabang saraf yang masuk ke canalis spinalis. Tindakan pada lesi ini
dengan laminectomy untuk komponen
intraspinal. Untuk komponen extraspinal dilakukan transforaminally jika tumor kecil dan letaknya di tengah, tetapi jika lesi besar maka diperlukan pemisahan lebih ke samping. Untuk lesi besar extraspinal daerah cervical maka dibuang dari arah lateral atau modifikasi secara hati-hati agar tidak merusak arteri vertebralis.37 Lesi cervicothoracic dibuang menggunakan cara posterior subscapular ke plexus brachialis. Lesi pada daerah thoracic diangkat dengan menggunakan cara costotransversectomy, lateral extracavitary, atau transthoracic intrapleural atau extrapleural bergantung pada ukuran lesi.38 Lesi lumbal biasanya
menggunakan cara retroperitoneal.
BAB 20 NEUROFIBROMATOSIS
293 anak- anak dengan NF1 terjadi
scoliosis,
penderita harus diperiksa klinis dan
radiologis secara serial terhadap
deformitas yang progresif setelah
operasi.40
Pertumbuhan craniofacial
neurofibroma secara umum bersifat plexiform yang melibatkan saraf perifer muka, orbita, dan cranial base. Lesi facial secara umum diterapi oleh bedah plastik karena dapat membahayakan nervus facialis akibat penekanan tumor atau kerusakan selama reseksi tumor.
Pertumbuhan craniofacial
neurofibroma secara umum bersifat plexiform yang melibatkan saraf perifer muka, orbita, dan cranial base. Lesi facial secara umum diterapi oleh bedah plastik karena dapat membahayakan nervus facialis akibat penekanan tumor atau kerusakan selama reseksi tumor. Lesi orbita merupakan tantangan secara kosmetik maupun fungsinya karena dapat
menyebabkan proptosis, divergent
strabismus, dan membahayakan penglihatan. Lesi ini juga dapat meluas secara intrakranial ke sinus cavernosus.41 Lesi ini jarang dapat direseksi total tanpa mengorbankan bola mata, maka pilihan operasi dilakukan pada penderita dengan hilangnya penglihatan dan pertumbuhan
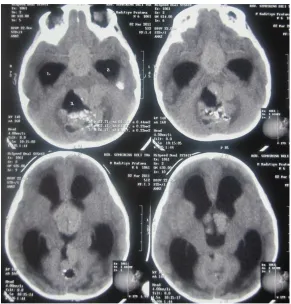
Gambar 3. Tampak lesi hiperdens pada CPA kiri yang menekan hemisfer serebellum dan CPA dengan defek pada os occipitalis kiri.
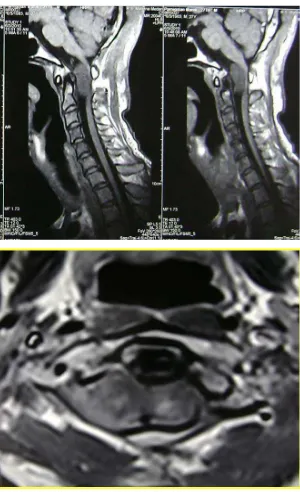
Gambar 4. MRI cervical. a. potongan sagital b. potongan aksial
Tampak massa tumor intradural extramedullary
daerah cervical medullary yang meluas
ke-paraspinal kanan melalui foramina intervertebralis C2 kanan.
tumor yang progresif. Penderita dengan penglihatan yang baik, fungsi stabilisasi selalu dapat dicapai dengan reseksi sebagian tumor karena sisa lesi
kadang-kadang tidak tumbuh kembali.
Penanganan visceral
neuroneurofibroma juga merupakan tantangan karena lesi ini biasanya bersifat plexiform yang tidak dapat dilakukan reseksi total. Reseksi subtotal selalu
diindikasikan untuk mengurangi
gangguan pulmonary (untuk lesi
intrathoracic yang besar), upper airway obstruction (dari parapharyngeal neurofibroma), abdominal discomfort, atau spinal cord compression (tumor mendorong komponen paravertebral dari transforaminal). Tumor seperti plexiform neurofibroma yang tidak direseksi cocok untuk percobaan kemoterapi.
Gambaran Radiologis
Karakteristik gambaran CT scan kepala pada NF1 selalu tampak pada suatu optic glioma, termasuk pembesaran fusiform dari saraf optic, tractus opticus, dan atau optic chiasma. Dalam hal ini terdapat juga perubahan bentuk canalis opticus dan sphenoid wing medialis. Sphenoid wing dysplasia biasanya berhubungan dengan
neurofibroma plexiform dan
bupthalmos.42 Dengan gambaran MRI otak, glioma saraf optic mudah tampak dengan pembesaran dari saraf optic, chiasma, dan/atau tractus opticus. Glioma optic pada penderita NF1 dapat ditemukan tanpa gejala hingga 20%. Pada tumor yang luas, T1-weighted images selalu di bawah estimasi, sedangkan T2-weighted images memberi representasi yang baik terhadap daerah yang terlibat. Brain stem glioma dengan pemberian kontras dapat terjadi enhancement yang homogen atau heterogen.43 Tumor parenchyma memunyai predileksi pada thalamus dan basal ganglia yang timbul
sebagai lesi yang panjang pada T2 dengan
variasi enhance setelah pemberian
gadolinium. Foci yang non-enhance dari T2 yang memanjang dalam nucleus deep gray dan white matter tampak vakuolisasi myelin. Ini paling sering pada globus pallidus, cerebellum, dan brain stem, kapsula interna, centrum semiovale, dan corpus callosum dengan insiden hingga 60% pada penderita NF1.
CT scan Schwannoma pada kepala memberikan gambaran suatu massa bulat atau ovoid pada extra-axial. Pada CT scan tanpa kontras ditemukan isomild hypodense, kecuali pada kista atau
perdarahan. Meningioma merupakan
massa extra-axial yang berhubungan dengan dural tail. Pada CT scan tanpa kontras ditemukan gambaran isodense dengan jaringan otak.43 Pada MRI, schwannoma isomild hypointense dibandingkan dengan jaringan otak pada T1-weighted images dan isohyperintense terhadap jaringan otak pada T2-weighted images. Setelah pemberian gadolinium,
tampak enhancement yang homogen
dibandingkan dengan kista atau
perdarahan yang cenderung heterogen. Lesi yang besar dapat menyebabkan
kompresi brain stem dan/atau
hydrocephalus. Mirip schwannoma, meningioma isointense terhadap gray matter pada T1 dan T2-weighted images.
Dengan pemberian gadolinium dapat
terjadi enhancement dengan gambaran kalsifikasi.43
Prognosis
BAB 20 NEUROFIBROMATOSIS
295 Penderita dengan NF1 34 kali lebih sering
mengalami keganasan jaringan ikat atau
tumor jaringan lunak dibandingkan
dengan individu non-NF1.9 Usia rata-rata dari onset gejala adalah tujuh belas tahun, sedangkan usia diagnosis NF2 rata-rata adalah dua puluh dua tahun. Prognosis penderita NF2 bervariasi sesuai dengan spektrum dari fenotipe. Tipe mutasi gen NF2 memengaruhi beratnya penyakit. Deteksi awal memberi keuntungan yang jelas pada penderita, seperti terlindungnya fungsi pendengaran. Diagnosis NF2
meningkatkan risiko berkembangnya
tumor CNS, seperti schwannoma,
meningioma, glioma, dan neuroma yang dapat melibatkan otak, saraf kranial, atau spinal cord.
Biologi Molekular NF1
NF1 merupakan suatu prototypical
hereditary tumor syndrome dari CNS. Sekitar 50% penderita adalah mutasi sporadic atau Inherited autosomal dominant. Efek terhadap individu berupa kelainan kulit dan merupakan predisposisi terjadinya berbagai tumor jinak dan ganas. Efek tumor terhadap CNS antara lain neurofibroma, malignant peripheral nerve sheath tumor, dan pilocytic astrocytoma (terutama jaras optic).44
Sedangkan non CNS malignancy
termasuk pheochromocytoma dan chronic myeloid leucemia pada remaja.
NF1 merupakan mutasi germline dari gen NF1 pada kromosom 17q11.2. NF1 adalah encode untuk suatu protein yang sangat besar dengan 57 exon yang menyebar lebih dari 350 kb dari genomic DNA. Lebih dari 200 perbedaan mutasi telah diobservasi pada penderita dengan NF1.45 Daerah NF1 coding biasanya berisi tiga gen lain dalam satu dari intronnya, yaitu EV12A, EV12B, dan OMGP.44 Peranan rangkaian gen NF1 dalam menentukan fenotipe NF1 adalah
tidak pasti. Protein encode melalui gen NF1 neurofibromin memunyai persamaan
dengan anggota Ras guanosine
triphosphatose-activating protein (Ras-GAP) family. Ras-GAP protein mengatur proto-oncogenic Ras. Mutasi aktivasi Ras ditemukan sekitar 30% dari semua kanker pada manusia.44 Pada berbagai tumor lain, aktivitas jaras Ras bertambah bila Ras
aktif berikatan dengan guanosine
triphosphate (GTP). Fungsi protein Ras-GTP untuk membuang Ras-GTP dari Ras, dan untuk mengikat guanosine diphosphate (membuat Ras menjadi tidak aktif).
Hilangnya fungsi protein Ras-GAP
cenderung meningkatkan kadar Ras-GTP, dan merupakan signal melalui jaras Ras. Signal Ras meninggi melalui signal receptor membrane-bound tyrosine kinase, seperti EGFR dan PDGFR.
Aktivitas Ras cenderung bersifat mitogenic melalui aktivasi Raf, MAPKK (mitogen activated protein kinas kinase), dan MAPK (mitogen activated protein kinase). Malignant peripheral nerve sheath tumor (MPNST) pada penderita
dengan NF1 tidak menunjukan
peningkatan kadar neurofibromin dan
Ras-CTP dibandingkan dengan
schwannoma pada penderita non NF1.46
Kemungkinan neurofibromin juga
memunyai fungsi supresi tumor yang tidak bergantung pada Ras.44 Beberapa tumor memunyai gen NF1 yang normal
pada tingkat DNA, yaitu bukan
merupakan neurofibromin yang
Encode gen NF1 untuk protein
dengan 2818 asam amino yang
dihubungkan dengan neurofibromin
tampak pada sel-sel Schwann,
oligodendrocyte, dan neuro-neuron yang bekerja sebagai gen supresi tumor. Protein berisi segment asam amino yang besar dan memperlihatkan persamaan terhadap fungsi domain dari protein aktivasi p21ras-GTPase. p21ras-GTPase inaktivasi oncogen p21ras dengan merangsang aktivitas GTPase, sehingga mengubah bentuk aktif dari p21ras menjadi bentuk
tidak aktif. Mutasi neurofibromin
cenderung rendah atau tidak terdapat ekspresi yang merupakan aktivasi p21ras, dan kemungkinan sejumlah kelainan fenotype yang tampak pada NF1,
termasuk tumor jinak dan ganas.
Transformasi keganasan terjadi dengan perubahan genetic tambahan, seperti mutasi p53. Terdapat kesan bahwa kelainan signal molecular. Pada dasarnya penderita NF1 tidak mau belajar dari pengalaman yang pernah terjadi. 48
NF2
Vestibular Neurilemoma
Tumor saraf kranial ke delapan terjadi terutama pada penderita dengan NF2 dan selalu simptomatik pada remaja. Anak-anak dengan catarac posterior atau tumor multiple spinal cord atau saraf perifer tanpa adanya Lisch nodule atau café-au-lait macules tetap harus dipikirkan tentang diagnosis NF2, dan dilakukan MR scan kepala untuk manifestasi intrakranial NF2.
Walaupun terdapat kesepakatan bahwa pemeriksaan pendengaran secara periodik dan MR scan pada vestibular neurilemoma untuk evaluasi penderita dengan NF2, namun indikasi dan teknik terapi untuk lesi ini masih kontroversi.
Penanganan primer, yaitu untuk
melindungi fungsi pendengaran tetapi
untuk mencapai hasil akhir masih belum pasti. Penderita dengan lesi yang besar dan menekan batang otak harus dilakukan operasi reseksi. Penderita dengan tumor yang kecil tanpa gejala belum terdapat
tindakan yang optimal. Beberapa
rekomendasi untuk mencoba reseksi
radikal tumor karena cara ini
memungkinkan untuk menjaga fungsi pendengaran setelah operasi. Terapi ini dapat menyebabkan tuli pada telinga kontralateral yang merupakan risiko jangka panjang. Reseksi tumor selalu dikombinasi dengan memasang implan pada cochlear atau auditory brainstem
yang setidaknya menjaga fungsi
pendengaran pada penderita dengan tumor bilateral yang dilakukan intervensi operasi.49
Stereotactic radiosurgery merupakan alternatif tindakan terapi.
Belum dapat dipastikan apakah
pemeliharaan fungsi pendengaran jangka panjang lebih baik daripada dengan reseksi terbuka. Hilangnya pendengaran dapat terjadi setelah radiosurgery, tetapi cara ini berkembang lambat sekitar satu sampai dua tahun kemudian. Cara lain yang dianjurkan pada penderita dengan tumor bilateral, yaitu reseksi lesi secara subcapsular dengan membiarkan sejumlah kecil tumor yang melekat pada
saraf facial dan acoustic untuk
meminimalisasi kerusakan saraf. Karena penanganannya yang kompleks, maka cara supraregional dianjurkan untuk mengoptimalkan hasil yang fungsional.
Intrakranial Meningioma dan
Nonvestibular Cranial Nerve
Neurilemoma
Lebih dari 30% penderita dengan NF2
memunyai multiple nonvestibular
BAB 20 NEUROFIBROMATOSIS
297 yang lebih dari 95% pada populasi
penderita ini. Walaupun memberi kesan bahwa subgrup penderita dengan NF2
memiliki risiko tinggi terhadap
multicenter intrakranial neurilemoma dan meningioma,50 tidak semua heterogenitas fenotipe dapat menerangkan perbedaan inherited pada susunan mutasi NF2 karena susunannya yang bervariasi dari pertumbuhan tumor memunyai catatan diantara penderita dengan mutasi identik dan juga diantara keduanya.51 Yang penting pemeriksaan penderita NF2 saat ini tidak memungkinkan untuk melakukan prediksi apakah penderita akan menderita multipel intrakranial tumor, dan tidak
adanya hubungan simpulan
genotype/fenotype sehingga terhadap semua penderita dilakukan pemeriksaan scan kepala secara periodic.
Banyak pendapat mengenai terapi vestibular neurilemoma dengan yang lesi lain, seperti intrakranial neurilemoma dan meningioma. Lesi ini memperlihatkan sifat biologis yang tidak dapat diprediksi. Beberapa tumor memunyai interval tetap atau tidak tumbuh meluas sedangkan yang lain cepat pertumbuhannya. Penderita
selalu mengalami perkembangan
intrakranial multipel neurilemoma dan meningioma, dan umumnya disiapkan intervensi operasi untuk lesi besar yang menyebabkan kompresi saraf dan yang memperlihatkan pertumbuhan progresif. Dengan scan dan diikuti pemeriksaan klinisnya, tumor ini jarang membesar
hingga menyebabkan kerusakan
neurologis yang ireversibel atau
memerlukan reseksi.
Intraparenchymal Glioma
Penderita dengan NF2 sering mengalami tumor glial intrinsic pada otak tetapi jarang ditemukan pada NF1. Sebaliknya, tumor intraspinal intramedullary lebih sering pada NF2.52 Pada observasi
penderita dengan NF1 ditemukan bahwa
penderita ependymoma lebih sering
daripada astrocytoma. Secara umum
ependymoma memiliki batas yang jelas sehingga memungkinkan reseksi total
pada lesi yang besar, dan yang
menunjukan gejala progresif.53
Penanganan setelah operasi menyerupai tumor spinal cord intramedullary.
Extracranial Neurilemoma dan
Meningioma
Penderita degan NF2 sering
memperlihatkan pertumbuhan intraspinal yang merupakan kombinasi nerve sheath tumor dan meningioma. Tumor ini tumbuhnya lambat dan bersifat menekan serta menginvasi struktur sekitar saraf, sehingga sering ditemukan lesi besar tanpa gejala selama beberapa tahun sebelum menimbulkan gejala klinis spinal cord. Lebih dari 90% tumor daerah spinal canal tanpa disertai defisit neurologis. Penderita dapat memperlihatkan fungsi neurologis yang cepat memburuk dengan pertumbuhan tumor yang kecil dan dianjurkan reseksi tumor pada penderita
dengan gambaran radiologis yang
memperlihatkan kompresi spinal cord walapun tanpa gejala. Nerve sheath tumor pada NF2 biasanya adalah neurilemoma,33 dan dengan neurofibroma yang khas pada NF1, lesi extracranial berbatas tegas, dan timbul dari single nerve fascicle yang selalu dapat dikorbankan dengan minimal neurologis morbidity sehingga lebih baik dilakukan reseksi total.34 Secara umum meningioma dapat dibuang tanpa reseksi berbagai elemen saraf. Sebagian kecil penderita dengan neurofibroma memiliki kesulitan yang sama dengan NF1.
Penderita perlu untuk membuang
Biologi Molekular NF2
NF2 merupakan suatu autosomal
dominant, hereditary dan tumor CNS syndrome yang dapat menjadi schwannoma, meningioma, ependymoma, dan tumor-tumor lain yang jarang. Semua family NF2 mengalami mutasi pada tumor supresi gen pada kromosom 22q12 yang
memiliki 595 asam amino dan
menghasilkan protein yang disebut merlin dan schwanomin.55 Protein merlin memperlihatkan homolog dengan anggota protein 4.1 superfamily dari protein-protein termasuk 4.1, talin, moesin, ezrin, dan radixin. Anggota protein 4.1 superfamily mengikat ke dua membran sel dan cytoskeleton yang mungkin terlibat dalam signal antara permukaan sel dan cytoskeleton. Lokasi merlin pada membran sel bekerja sebagai ikatan membrane-cytoskeletal. Walaupun usaha telah dilakukan untuk mengidentifikasi fungsinya, tetapi masih belum jelas keterlibatan berbagai jaras signal merlin-regulated sebagai kunci dari perkembangan NF2 yang berhubungan
dengan tumor.56 Mutasi cenderung
menyebabkan hilangnya ekspresi merlin dan merupakan defek gen paling sering pada meningioma. Total 50-60% dari semua meningioma spontan, dan NF2 berhubungan dengan meningioma yang
mengalami mutasi pada gen NF2.
Schwannoma yang disebabkan oleh hilangnya ekspresi merlin dan hanya 29-38% dari ependymoma memperlihatkan perubahan ekspresi merlin.57 Mutasi dari gen NF2 terjadi tidak hanya pada tumor yang berhubungan dengan NF2, tetapi juga mencapai 30% dari melanoma, dan 41% dari mesothelioma.58 Masih belum jelas mengapa mutasi NF2 cenderung untuk membentuk vestibular schwannoma bilateral. Mutasi merlin menyebabkan defek pada membrane-cytoskeleton yang
merusak pertumbuhan dengan
penghambatan signal dari permukaan sel dan menghasilkan fenotype tumor. NF2 memperlihatkan berbagai gejala klinis. Beberapa penderita memunyai tumor multipel pada usia muda, dan yang lain memunyai gejala minimal pada usia tua. Penderita dengan terpotongnya mutasi dari NF2 memunyai fenotype lebih berat daripada perubahan single codon dan menunjukan fenotype yang berhubungan dengan NF2 family. Sindroma family
disebut juga schwanomatosis yang
memberi efek pada individu dengan multiple schwannoma tetapi bukan schwannoma vestibular.59 Tumor pada penderita ini selalu khas dengan mutasi NF2.
Hilangnya heterozygosity pada kromosom 22q sering pada meningioma, ependymoma, dan schwannoma. Ekspresi merlin yang hilang atau berkurang selalu ditemukan pada tumor sporadic ini.60
Mutasi gen NF2 ditemukan pada
ependymoma spinal, tetapi bukan ependymoma pada ventrikel IV dan anak-anak. Hal ini sesuai dengan distribusi
penderita ependymoma dengan NF2.
Walaupun banyak astrocytoma
memperlihatkan hilangnya heterozygosity pada kromosom 22q, tetapi mutasi gen NF2 tidak ditemukan pada astrocytoma. Hal ini memberi kesan ke dua gen supresi tumor pada chromosom 22q terlibat dalam tumorgenesis astrocytoma.61 Mutasi gen NF2 ditemukan mencapai 30-60% dalam bentuk sporadic meningioma. Mutasi NF2 lebih sering terjadi pada fibroblastic dan meningioma transitional tetapi jarang terjadi pada meningioma meningothelial.
Hai ini memberi kesan adanya
kemungkinan heterogeneity molecular pada tumor ini.
Kelainan neurofibromatosis ke
tiga telah dibedakan sebagai
BAB 20 NEUROFIBROMATOSIS
299 cranial selain saraf vestibular.
Kelainannya pada gen kromosom 22
dekat locus NF2 tetapi timbul dari NF2.62
Terdapat perdebatan apakah
hemangiopericytoma dural merupakan varian dari meningioma atau keseluruhan
tumor. Hemangiopericytoma tidak
memunyai mutasi pada gen NF seperti meningioma, tetapi ditemukan hilangnya locus CDKN2A dan tidak terdapat pada meningioma. Ini memberikan bukti
tambahan bahwa hemangiopericytoma
adalah meningioma.63 Tidak semua
sporadic meningioma atau schwannoma memunyai mutasi NF2. Diantara sporadic meningioma dan schwannoma yang tidak
memunyai mutasi NF2 terdapat
peningkatan proteolysis dari merlin melalui calpain dan fungsi dari merlin tidak aktif.64
Terapi
Penderita NF1 secara keseluruhan harus dilakukan pemeriksaan fisik termasuk
lapangan pandang. Dilakukan
pemeriksaan MRI kepala dan tulang belakang. Walaupun terdapat massa, terapi dilakukan apabila ditemukan gejala akibat lesi.65 Kebanyakan glioma jaras optic yang berhubungan dengan NF1 tidak menimbulkan gejala dan beberapa mengalami regresi spontan.66 Patologis tumor ini biasanya pilocytic astrocytoma WHO grade I, dengan klinis lebih lambat dibandingkan dengan penderita non-NF1. Peranan operasi pada penderita glioma jaras optic masih kontroversial. Intervensi operasi dilakukan pada lesi tunggal yang menimbulkan gejala dan mengganggu penderita. Operasi bermanfaat apabila terdapat gejala peningkatan tekanan
intrakranial, efek massa, atau
hydrocephalus.67
Pertumbuhan tumor yang cepat lebih sering terletak pada hypothalamus dan chiasma. Harus dilakukan dengan segera
operasi reseksi untuk melindungi
penglihatan dan mengurangi efek massa.68 Terapi radiasi kurang bermanfaat pada penderita NF1, karena efek samping terhadap perkembangan neurovascular,
endokrin, dan neuropsychological,
disamping risiko tinggi berkembangnya menjadi keganasan sekunder.69 Terapi radiasi pada glioma jaras optic yang progresif memunyai risiko menyebabkan suatu tumor sekunder CNS, namun tidak ditemukan pada penderita dengan tumor yang sporadic. Pertumbuhan yang lambat dari glioma jaras optic tampak pada foto serial, dan bila disertai gejala dapat dilakukan sistemik kemoterapi. Penderita
NF1 dengan low-grade glioma yang
dilakukan kemoterapi secara keseluruhan lebih baik dibandingkan dengan penderita non-NF1.70 Penderita dilakukan terapi jika ditemukan satu atau lebih dari kriteria sebagai berikut:
1. Ukuran tumor bertambah >25% 2. Papilledema
3. Hilangnya penglihatan 4. Proptosis yang progresif, atau
5. Diameter saraf optic yang
bertambah >2 mm
Operasi selalu dilakukan untuk
neurofibroma plexiform yang
menyakitkan, tetapi pengembangan
biotechnology yang baru sedang dalam penelitian.71 Penanganan terbaik untuk schwannoma masih kontroversi.
Kemungkinan gejala hilangnya
pendengaran akibat kegagalan
penanganan konservatif. Operasi
vestibular schwannoma masih menjadi tantangan, dan selalu dilakukan oleh multidisiplin, seperti ahli bedah saraf dan
neuro-otologis. Radiosurgery lebih
efektif untuk terapi tumor NF2, dan angka perlindungan pendengaran lebih baik (sekitar 43%). Erlotinib adalah suatu inhibitor EGFR yang efektif dalam terapi vestibular schwannoma yang progresif.73
Penggunaan EGFR inhibitor ini
berdasarkan atas pemeriksaan, bahwa merlin berhubungan dengan EGFR melalui tingkatan protein dan pencegahan signal. Pada penderita dengan NF2 dan mutasi merlin, jaras ini tidak dapat diatur ke bawah.74
Tinjauan Pustaka
1. Huson SM, Hughes RAC, eds. The Neurofibromatoses. A Pathogenetic and Clinical Overview. New York: Chapman and Hall; 1994
2. Korf BR. Clinical features and pathobiology of neurofibromatosis 1.
Journal of Child Neurology
2002;18(8):573-577;discussion 602-604,646-651
3. Debella K, Szudek J, et al. Use of the national institudes of health crtiteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000;105(3 Pt 1):608-614
4. Xu G, O’Connell P, Viskochil D, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990;62:599-608
5. Basu TN, Gutmann DH, Fletcher JA, et al. Aberrant regulation of ras proteins in malignant tumors cells
from type 1 neurofibromatosis
patients. Nature 1992;356:713-715 6. Matsui I, Tanimura M, Kobayashi N,
et al. Neurofibromatosis type 1 and
childhood cancer. Cancer
1993;72:2746-2754
7. Listernick R, Charrow J.
Neurofibromatosis-1 in childhood .
Advances in Dermatology
2004;20:75-115
8. van Engelen SJ, Krab LC, et al. Quantitative differentiation between healthy and disordered brain matter in patients with neurofibromatosis type 1 using diffusion tensor imaging.
AJNR. American Journal of
Neuroradiology 2008;29(4):816-822
9. Rasmussen SA, Yang Q, et al.
Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. American Journal of Human Genetics 2001;68(5):1110-1118
10. Uppal S, Coatesworth AP.
Neurofibromatosis type 2,
International Journal of Clinical Practice 2003;57(8):698-703
11. Dow G, Biggs N, et al. Spinal tumors in neurofibromatosis type 2. Is emerging knowledge of genotype predictive of natural history? Journal
of Neurosurgery. Spine
2005;2(5):574-579
12. Martuza RL, Eldridge R.
Neurofibromatosis 2 (bilateral) acoustic neurofibromatosis). N Engl J Med 1988;318:684-688
13. Rouleau GA, Seizinger BR,
Wertelecki W, et al. Flanking markers bracket the neurofibromatosis type 2 (NF2) gene on chromosome 22. Am J Hum Genet 1990;46:323-328
14. Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin, ezrin, radixin-like gene is a candidate for
the neurofibromatosis 2 tumor
suppressor. Cell 1993;72:791-800 15. Ingordo V, D’Andria G, Mendicini S,
et al. Segmental neurofibromatosis: is it uncommon or underdiagnosed?
(letter). Arch Dermatol
1995;131:959-960
16. Isenberg JS, Mayer P, Butler W, et al.
Multiple recurrent benign
schwannoma of deep and superficial nerves of the upper extremity: a new
BAB 20 NEUROFIBROMATOSIS
301 neurofibromatosis. Ann Plast Surg
1994;33:659-663
17. Di Simone RE, Berman AT,
Schwentker EP. The orthopedic
manifestation of neurofibromatosis: a clinical experience and review of the literature. Clin Orthop 1988;230:277-283
18. Ward BA, Harkey HL, Patent AD, et
al. Severe cervical kyphotic
deformities in patients with plexiform
neurofibromas: case report.
Neurosurgery 1994;35:960-964 19. Huson SM, Harper PS, Compston
DAS. Von Recklinghausen
neurofibromatosis. A clinical and population study in south-east Wales. Brain 1988;111:1355-1381
20. Stiller CA, Chessels JM, Fitchett M. Neurofibromatosis and childhood leukemia-lynphoma: a population-based UKCCSG study. Bs J Cancer 1994;70:969-972
21. Mulvihill JJ. Malignancy
epidemiologically associated cancers. In: Huson SM, Hughes RAC, eds.
The neurofibromatosis: A
Pathogenetic and Cliniocal Overview. New York: Chapman and Hall; 1994:305-315
22. Mulvihill JJ, Parry DM, Sherman Jl,
et al. Neurofibromatosis 1
(Recklinghausen disease) and
Neurofibromatosis 2 (bilateral
acoustic neurofibromatosis). An update. Ann Intern Med 1990;113:39-52
23. Itoh T, Magnaldi S, White RM, et al.
Neurofibromatosis type-1. The
evolution of deep gray and white
(unidentified bright objects) and
lower IQs in children wirh
neurofibromatosis-1. Am J Med
Genet. (Neuropsych Genet)
1996;67:98-102
25. Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol 1994;38:427-452
26. Sutton LN, Molloy PT, Seryak H, et
al. Long-term outcome of
hypothalamic/ chiasmatic
astrocytomas in children treated with conservative surgery. J Neurosurg 1995;83:583-589 complications of radiation therapy for pediatric brain tumors. Pediatr Neurosurg 1992;18:207-217
29. Packer RJ, Ater J, Allen J, et al.
Caoboplatin and vincristine
chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg 1997;86:747-754
30. Ferner RE, Chaudhuri R, Bingham J, et al. MRI in neurofibromatosis 1. The nature and evolution of increased T2 weighted lesions and their
relationship to intellectual
impairment. J Neurol Neurosurg Psychiatry 1993;56:492-495
31. Mapstone TB. Neurofibromatosis and central nervous system tumors in childhood. Neurosurg Clin North Am 1992;3:771-779
32. Pollack IF, Pang D, Albright AL. The long-term outcome in children with
late-onset aqueductal stenosis
neurofibromatosis type 1 and 2. J Neurosurg 1991;74:248-253
34. Donner TR, Voorhies RM, Kline DG. Neural sheath tumors of major nerves. J Neurosurg 1994;81:362-373
35. Kline DG, Judice DJ. Operative management of selected brachial
plexus lesions. J Neurosurg
1983;58:631-649
36. Bruckner HW, Gorbaty M, Lipsztein R, et al. Treatment of large
high-grade neurofibrosarcoma with
concomitant vinblastine, doxorubicin, and radiotherapy. Mt Sinai J Med 1992;59:429-432
37. George B, Lot G. Neurinoma of the first two cervical nerve roots: a series
of 42 cases. J Neurosurg
1995;82:917-923
38. McKormick PC. Surgical
management of dumbbell and
paraspinal tumors of the thoracic and
lumbar spine. Neurosurgery
1996;38:67-75
39. Schultheiss R, Gullotta G. Resection of relevant nerve roots in surgery of spinal neurinoma without persisting neurological deficit. Acta Neurochir (Wien) 1993;122:91-96 Management of tumours involving
the cavernous sinus Acta
Neurochirurgica (Wien) 1991;53:101-112(supplement)
42. Kornreich L, Blaser S, et al. Optic pathway glioma: correlation of imaging findings with the presence of neurofibromatosis. AJNR. American
Journal of Neuroradiology
2001;22(10):1963-1969
43. Fischbein NJ, Dillon WP, et al. Teaching atlas of brain imaging. Thieme, New York 2000
44. Feldkamp MM, Gutmann DH, Guha A. Neurofibromatosis type 1 : Piecing pizzle together. Can J Neurol Sci 1998;25:181-191
45. Pros E, Gomez C, et al. Nature and mRNA effect of 282 different NF1 point mutationas: focus on splicing
alterations. Human Mutation
2008;29(9):E173-E193
46. Guha A, Lau N, Huvar I, et al. Ras-GTP levels are elevated in human
NF1 peripheral nerve tumors.
Oncogene 1996;12:507-513
47. Skuse Gr, Cappione AJ, Sowden M, et al. The neurofibromatosis type 1 messenger RNA undergoes base-modification RNA editing. Nucleic Acids Res 1996;24:478-486
48. Szudek J, Birch P, et al. Associations
of clinical features in
neurofibromatosis 1 (NF1). Genetic Epidemiology 2000;19(4):429-439 49. Hulka GF, Bernard EJ, Pillsbury HC.
Cochlear implantation in a patient after removal of an acoustic neuroma.
The implications of magnetic
resonance imaging with gadolinium
on patient management. Arch
Otolaryngol Head Neck Surg
1995;121:465-468
50. Parry DM, Eldridge R, Kaiser-Kupfer MI, et al. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet 1994;52:450-461
51. Baser ME, Ragge NK, Riccardi VM, et al. Phenotypic variability in
monozygotic twins with
neurofibromatosis 2. Am J Med Genet 1996;64:563-657
52. Mautner V-F, Tatagiba M, Guthoff R, et al. Neurofibromatosis 2 in the pediatric age group. Neurosurgery 1993;33:92-96
BAB 20 NEUROFIBROMATOSIS
clinical spectrum of
neurofibrometosis 2. Neurosurgery 1996;38:880-886
55. Louis DN, Ramesh V, Gusella JF.
Newuropathology and molecular
genetic of neurofibromatosis 2 and
related tumors. Cancer Res
1995;5:163-172
56. Scoles DR. The merlin interacting protein reveal multiple targets for
NF2 therapy. Biochemica et
Biophysica Acta 2008;1785(1):32-54 57. Rajaram V, Gutmann DH, et al.
Alterations of protein 4.1 family members in ependymomas: a study of
84 cases. Modern Pathology
2005;18(7):991-997
58. De Vitis LR, Tedde A, et al. Analysis of the fibromatosis type 2 gene in
different human tumors of
neuroectodermal origin. Human
Genetics 1996a,b;97(5):638-641
60. Gutmann DH, Giordano MJ, Fishback BS, et al. Loss of merlin expression in
sporadic meningiomas,
ependymomas, and schwannomas. Neurology 1997;49:267-270
61. Rubio MP, Correa KM, Ramesh V, et al. Analysis of the neurofibromatosis 2 gwene in human ependymomas and
astrocytomas. Cancer Res
1994;54:45-47
62. MacCollin M, Willett C, et al. Familial schwannomatosis: exclusion of the NF2 locus as the germline event. Neurology 2003;60(12):1968-1974
63. Ono Y, Veki K, Joseph JT, et el. Zygous deletions of the CDKN2/p16 gene in dural hemangiopericytomas. Acta Neuropathol 1996;91:221-225 64. Kimura Y, Koga H, Araki N, et al.
The involvement of calpain
dependent proteolysis of the tumor
suppressor NF2 (merlin) in
schwannomas and meningiomas. Nat Med 1998;4:915-922
65. Pollack IF, Shultz B, et al. The management of brain stem gliomas in patients with neurofibromatosis 1. Neurology 1996;46(6):1652-1660
66. Parsa CF, Hoyt CS, et al.
Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Archives of Ophthalmology 2001;119(4):516-529 67. Astrup J. Natural history and clinical
management of optic pathway glioma. British Journal of Neurosurgery 2003;17(4):327-335
68. Listernick R, Ferner RE, et al. Optic
pathway gliomas in
neurofibromatosis-1: controversies and recommendations. Annals of Neurology 2007;61(3):189-198 69. Sharif S, Ferner R, et al. Second
primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy.
Journal of Clinical Oncology
2006;24(16):2570-2575
70. Gururangan S, Cavazos CM, et al. Phase II study of carboplatin in children with progressive low-grade gliomas. Journal of Clinical Oncology 2002;20(13):2951-2958
71. Babovic-Vuksanovic D, Widemann BC, et al. Phase I trial of pirfenidone in children with neurofibromatosis 1
and plexiform neurofibromas.
Psdiatric Neurology 2007;36(5):293-300
72. Myrseth E, Pedersen PH, et al.
schwannomas. Why, when, and how?
Acta Neurochirurgica
2007;149(7):647-660; discussion 660 73. Plotkin SR, Singh MA, et al.
Audiologic and radiographic response
of NF2 related vestibular
schwannoma to erlotinib therapy. Nature clinical practice. Oncology 2008;5(8):487-491
74. Curto M, Cole BK, et al.
Contact-dependent inhibition of EGFR