Dav i d
David Publishing Company www.davidpublishing.com P u b l i s h i n g Dav i d
Journal of Life Sciences
Publication Information
Journal of Life Sciences is published monthly in hard copy (ISSN 1934-7391) and online (ISSN 1934-7405) by David Publishing Company located at 9460 TELSTAR AVE SUITE 5, EL MONTE, CA 91731, USA.
Aims and Scope
Journal of Life Sciences, a monthly professional academic journal, covers all sorts of researches on molecular biology, microbiology, botany, zoology, genetics, bioengineering, ecology, cytology, biochemistry, and biophysics, as well as other issues related to life sciences.
Editorial Board Members
Dr. Stefan Hershberger (USA), Dr. Suiyun Chen (China), Dr. Farzana Perveen (Pakistan), Dr. Francisco Torrens (Spain), Dr. Filipa João (Portugal), Dr. Masahiro Yoshida (Japan), Dr. Reyhan Erdogan (Turkey), Dr. Grzegorz Żurek (Poland), Dr. Ali Izadpanah (Canada), Dr. Barbara Wiewióra (Poland), Dr. Valery Lyubimov (Russia), Dr. Amanda de Moraes Narcizo (Brasil), Dr. Marinus Frederik Willem te Pas (The Netherlands), Dr. Anthony Luke Byrne (Australia), Dr. Xingjun Li (China), Dr. Stefania Staibano (Italy), Dr. Wenle Xia (USA), Hamed Khalilvandi-Behroozyar (Iran).
Manuscripts and correspondence are invited for publication. You can submit your papers via Web Submission, or E-mail to [email protected] or [email protected]. Submission guidelines and Web Submission system are available at http://www.davidpublishing.com.
Editorial Office
9460 TELSTAR AVE SUITE 5, EL MONTE, CA 91731, USA Tel: 1-323-9847526, Fax: 1-323-9847374
E-mail:[email protected], [email protected]
Copyright©2013 by David Publishing Company and individual contributors. All rights reserved. David Publishing Company holds the exclusive copyright of all the contents of this journal. In accordance with the international convention, no part of this journal may be reproduced or transmitted by any media or publishing organs (including various websites) without the written permission of the copyright holder. Otherwise, any conduct would be considered as the violation of the copyright. The contents of this journal are available for any citation. However, all the citations should be clearly indicated with the title of this journal, serial number and the name of the author.
Abstracted / Indexed in
Database of EBSCO, Massachusetts, USA Chemical Abstracts Service (CAS), USA Cambridge Scientific Abstracts (CSA), USA
Chinese Database of CEPS, American Federal Computer Library center (OCLC), USA Ulrich’s Periodicals Directory, USA
Chinese Scientific Journals Database, VIP Corporation, Chongqing, China Universe Digital Library S/B, Proquest
Subscription Information
Price (per year): Print $520, Online $360, Print and Online $680.
David Publishing Company
9460 TELSTAR AVE SUITE 5, EL MONTE, CA 91731, USA Tel: 1-323-9847526, 323-410-1082; Fax: 1-323-9847374 E-mail: [email protected]
David Publishing Company www.davidpublishing.com
DAV ID P UBL ISH IN G
J LS
Journal of Life Sciences
Volume 7, Number 3, March 2013 (Serial Number 59)
Contents
Molecular Biology and Bioinformatics
219 Gene-Ontology Analysis on the Differentially Expressed Genes in Maize (Zea mays L.) Ear Rot
Guang-Sheng Yuan, Jian Gao, Zhi-Ming Zhang, Juan Du, Gui-Qing Mu and Guang-Tang Pan
227 Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction of Candidate Genes
Infected by SCMV in Zea mays Genome
Li Liu, Xiu-Jing He, Zhi-Ming Zhang, Mao-Jun Zhao and Guang-Tang Pan
236 BRCA1 Mutation Detection Using Fluorescent Hybridization Probes and Melting Curves
Safa R. Fitouri, Nouri B. Ermeli, Salah M. Bensaber, Mousa I. Jaeda, Ibrahim A. Mrema, Anton Hermann and Abdul M. Gbaj
244 Clustering and Expression Analysis of Chitinases in Maize and Rice
Kui Xiang, Wei-Tao Li, Xue-Wei Chen, Guang-Sheng Yuan, Wei-Lan Chen, Zhi-Ming Zhang, Ya-Ou Shen, Hai-Jian Lin and Guang-Tang Pan
252 Simulation of Spread of Infectious Diseases and Population Mobility in a Deterministic Epidemic Patch Model
Ariel Félix Gualtieri and Juan Pedro Hecht
Microbiology
259 Tests of Antibiotic Properties of Algerian Desert Truffle against Bacteria and Fungi
Samir Neggaz and Zohra Fortas
267 Detection of Delayed Hypersensitivity to Fonsecaea pedrosoi Metabolic Antigen (Chromomycin) in Healthy People in an Endemic Area
276 Inhibitory Effect of the Essential Oil from Hyptis suaveolens (L.) Poit on the Growth and Aflatoxins Synthesis of Aspergillus flavus
Ana Carolina Pessoa Moreira, Egberto Santos Carmo, Paulo Alves Wanderley, Evandro Leite de Souza and Edeltrudes de Oliveira Lima
Botany and Zoology
282 Effect of Some Bioproducts on Winter Mortality of Grafted Buds and the Number of Maiden
Fruit Trees Produced in an Organic Nursery
Zygmunt Stanisław Grzyb, Wojciech Piotrowski, Paweł Bielicki and Lidia Sas Paszt
289 Comparative Evaluation of NPK Fertilizer and Tithonia diversifolia Biomass in Sweet Pepper
(Capsicum annum) Production in Ado Ekiti, Nigeria
Ademiluyi Benson Oluwafemi
293 Sprinkler Irrigation and Soil Tillage Practices in Sugarcane Plantations as Influenced by Soil
Texture and Water Storage in Northern Ivory Coast
Crépin B. Péné, Souleymane N’Diaye and Chantal N’Guessan-Konan
302 Effects of a Brine Discharge over Bottom Polychaeta Community Structure in Chabahar Bay
Seyyed Mohammad Bagher Nabavi, Mohadese Miri, Babak Doustshenas, Ali Reza Safahieh and Mehran Loghmani
Interdisciplinary Researches
308 Real and Legal Nutritional Alternative (e.g. Application of Free Amino Acids) to Replace
Forbidden Doping Substances to Produce Excellent Sport Performance
Andras S. Szabo
313 The Bioclimate in the Steppe of Tlemcen (Oran, Western Algeria)
Assia Bekkouche, Fouzia Ayache and Mohammed Bouazza
322 Biology and Culture
Mar. 2013, Vol. 7, No. 3, pp. 219-226
Journal of Life Sciences, ISSN 1934-7391, USA
Gene-Ontology Analysis on the Differentially Expressed
Genes in Maize (
Zea mays
L.) Ear Rot
Guang-Sheng Yuan, Jian Gao, Zhi-Ming Zhang, Juan Du, Gui-Qing Mu and Guang-Tang Pan
Maize Research Institute, Sichuan Agricultural University, Ya’An 625014, China
Received: November 29, 2012 / Accepted: January 24, 2013 / Published: March 30, 2013.
Abstract: To better know FM (Fusarium moniliforme) induced genes in maize ear rot, GO (gene ontology) method was performed to analyze detail physiological functions in the defensive response after pathogen infection. This gene annotation system was widely used to investigate large numbers of genes involving in real active role or regulator in cell response. First of all, differentially expressed genes were isolated by using genechip platform at 96 h post-inoculation with FM in maize inbred Bt-1. In total, 482 differentially expressed unique genes were screened out in inbred Bt-1 when compared to mock-inoculated bract tissues. Then, each gene was annotated to define functional class by GO method. Finally, these large FM-responsive genes with significant differentially change were sorted into cellular component, molecular function and biological process with complicated network by molecular annotation system. The demonstrated information in the GO analysis could provide another view for understanding the molecular mechanism and indicate a deeply complicated network with gene function underlying disease development in the host tissue. The findings in this study provide important bases to probe the molecular processes, the alteration of metabolism and the immune mechanism upon the FM infection in maize.
Key words: Ear rot, genechip, Fusarium moniliforme, gene ontology, Zea mays.
1. Introduction
Fusarium ear rot, predominantly caused by FM (Fusarium moniliforme), F. proliferatum, and F.
subglutinans, is among the most destructive diseases for its decrease of grain yield in maize [1-3]. Especially,
a high incidence of ear rot occurs in the moist and humid regions of southwest China, as well as other regions with similar longitude in other countries [4, 5].
The symptom for Fusarium ear rot usually consists of a white or light pink mold on bracts or kernels [6].
Biochemical treatments and planting resistant maize inbred lines are the most common methods for controlling this disease. However, current resistant
inbred lines are only partially resistant, and severe outbreaks of ear rot can occur when climatic conditions
are favorable for the pathogen [7]. In addition, the
Corresponding author: Guang-Tang Pan, Ph.D., professor, research fields: plant genetics and breeding. E-mail: [email protected].
ingestion of FM infected grain can cause severe adverse effects in both humans and animals due to the
production of diverse and potent mycotoxins [8]. A variety of active defense mechanisms are known in
plants to protect them from microbial pathogen infection [9]. After specific recognition of a pathogen, the HR (hypersensitive responses) are induced in plants
to resist microbial pathogens. It has been proven that the most efficient way to control plant diseases is to
build up the most resistance in new cultivars. To this end, it is great important to breed efficient, broad-spectrum and stable-resistant cultivars with
resistant potential function in maize ear rot.
Within the past several years, considerable progress
has been made in investigation of the resistant system involved in maize ear rots infected by FM, including isolation of disease resistance genes, characterization
Gene-Ontology Analysis on the Differentially Expressed Genes in Maize (Zea mays L.) Ear Rot 220
responses [10-12]. In response to FM infection, the
PR-like proteins, chitinases, -1,3-glucanases and calcium-dependent protein kinase were overproduced in maize [13]. Moreover, the disease resistance genes
Hm1 and guanylyl cyclase-like protein (ZmGC1) were isolated closely involving in maize ear rot [14, 15]. Despite several resistant genes on FM infection
described in maize, their possible molecular mechanisms underlying activation of plant defense
responses are still unknown. It is widely accepted that plant disease resistances, such as to maize ear rot, are controlled by a multigene trait linking with QTL
(quantitative trait loci). In our previous works, we have isolated and mapped several QTLs on
chromosome using maize cultivars R15 (resistant) and Ye478 (susceptible) for resistance to FM ear rot [16]. Although QTLs mapping have advanced our
knowledge regarding the genetic mechanisms of disease resistance, the molecular processes and gene
regulation of the defense system relevant to maize ear rot remains poorly understood.
In this study, one objective was to use Gene
ontology method analyzing host gene expression changes in maize inbred lines: Bt-1, which is
completely resistant to FM. The inbred line has been investigated for many years for response to FM infection in southwest China. To better understand the
host genes involved in the maize defense response to ear rot, the authors examined gene expression changes
in bract tissue of resistant maize inbred line Bt-1 at the 4 day after inoculation with FM, using a whole-genome genechip. Results showed that 482
genes were specifically found in resistant line Bt-1. Finally, these large number genes were annotated to
different functional categories by Gene ontology. We found that the host genes can be involved in cellular component, molecular function, biological process
with complicated network to FM infection. Overall, the present study might help promote further
understanding of mechanisms underlying in maize defense against this pathogen.
2. Materials and Methods
2.1 Plant Materials and Inoculation Procedures
A resistant maize inbred line Bt-1 with high level
resistance to FM (Fusarium moniliforme), preliminary evaluated through many years for field trial, was used in this study. The line Bt-1 is derived
from the tropical germplasm with high resistance to
Fussrium ear rot and excellent agronomic characters
in maize. The spores of FM were cultured on PDA (potato dextrose agar) media for 15 days prior to collection for inoculations. Inoculum was prepared
by washing conidia from the cultures and diluting to a final concentration of approximately 1.0 × 106
spores/mL in water. Milky stage maize plants were inoculated with 3 mL on each bract by injection. The inoculated plants were grown under controlled
conditions at the Maize Research Institute of Sichuan Agricultural University.
2.2 Sampling and Affymetrix Chip Hybridization
To identify specifically expressed genes in response
to FM inoculation at the fourth day, genechip hybridization were performed using RNAs from the
independent FM-infected bract tissues and their controls. After HR (hypersensitive responses) occurring, the 96 h post-inoculation and mock-inoculation bract tissues
were sampled by collecting two independent biological replicates, each consisting of independent maize bract
tissue. Samples were frozen immediately in liquid nitrogen and sent to the Bioassay Laboratory of Capital Bio Corporation (Beijing, China) for cDNA synthesis,
labeling, hybridization to the maize Affymetrix GeneChip Maize Genome Array (Affymetrix, Santa
Clara, CA, USA), washing, and scanning. The genechip tool contains 17,555 probe sets representing 14,850 maize gene transcripts, and can provide a powerful
resource for characterizing the host response at the gene expression level in maize ear rot disease. Details of the
Gene-Ontology Analysis on the Differentially Expressed Genes in Maize (Zea mays L.) Ear Rot 221
products/arrays/specific/maize.affx. Both treated and control bract tissues were performed for two replicates.
2.3 Affymetrix Chip Data Analysis
Resulting Affymetrix data files are publically available from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under the
accession number GSE19501. Data analysis was performed using Affymetrix GeneChip Operating
SoftwareVersion 1.4 (GCOS) as described (Affymetrix Statistical Algorithms Reference Guide) [17]. Raw data was normalized by dividing each probe
set value by the median of that probe set from all samples, effectively centering the data around 1 and
enabling simple identification of differentially expressed genes. Statistical analysis was performed to identify genes that were differentially expressed in
FM-inoculated samples compared to mock-inoculated samples using analysis of variance (ANOVA, P <
0.05) across all replicates. Induction or repression of significantly differentially expressed genes was determined by dividing the raw signal value for each
replicate from FM-inoculated bract tissues by the average of the raw signal values from
mock-inoculated controls. Genes were then described as “up-regulated” or “down-regulated” if their change in expression was > 1.5-fold.
2.4 Annotation and Sequence Alignment
Annotation of gene sequences was performed by
searching the NCBI database (http://www.ncbi.nih.gov/) for homology sequences
using the BLASTx (Basic Local Alignment Search Tool X) algorithm. The putative physiological
functions of sequences were classified according to the gene ontology analysis.
3. Results and Discussion
3.1 FM-induced Genes Expression Changes in Bt-1
Using the GeneChip Maize Genome Array platform,
large-scale gene expression analysis was investigated
in maize bract tissue in resistant line Bt-1 at 96 h after inoculation with FM. In total, 482 unique genes were
found as be up-regulated more than 1.5-fold in resistant line Bt-1 (ANOVA, P < 0.05) when
compared to mock-inoculated bract tissues (Fig. 1). Further analysis of the 482 FM-induced genes identified in Bt-1 line indicated that 372 are already
annotated, since the remaining 110 unknown, based on the UniGene assignment (published data in
Ref. [18]). Bioinformatic analysis was undertaken to assign a description and functional categorization to the FM-induced genes.
3.2 Molecular Function Annotation of FM-induced
Genes in Bt-1
GO (Gene ontology) method was used for
functional classification to the 482 FM-induced genes by the Web Gene Ontology Annotation Plot and the results were plotted in Fig. 2. Using all genes in the
plant genome as background for significance testing, we found that these genes were involved in three
functional categories including cellular component, molecular function and biological process. Further classification of these differentially expressed genes
were significantly enriched in seven cellular component categories, five molecular function
categories, and nine biological process categories.
3.3 GO Annotation Involved in Cellular Component
The further research was to examine the function of the differentially expressed genes involved in cellular
component. It was found that quite there are a few genes associated with organelle in plant cellular
component, such as membrane, cytoplasm or nucleolus. As showed in Fig. 3, many genes were observed particularly involving in cellular plasma
membrane part. The result indicates that the interaction between host tissues and pathogen
occurred at cellular membrane surface first. The previous document showed that the host defense genes in cellular membrane could be stimulated firstly after
Gene-Ontology Analysis on the Differentially Expressed Genes in Maize (Zea mays L.) Ear Rot 222
Fig. 1 Cluster analysis on Affymetrix GeneChip maize genome array.
Fig. 2 Molecular annotation on differentially expressed genes from genechip data.
infection in plant, it is usually interacted with
membrane in cell, which has been proposed to orchestrate the establishment of different defensive
barriers against pathogen [19].
3.4 GO Annotation Involved in Molecular Function
In the GO analysis, many differentially expressed genes showed important physiological functions
associating with signal transduction, dehydrogenation oxidate, phosphorylation, protein kinase, transcriptional regulation, and ROS (reactive oxygen scavenging). The functional genes could provide deeply information on physiological responses during
FM inoculation (Fig. 4). Early literature illustrated that genes involving in signal transduction pathways may play different important roles in the host defense
CK1 CK2 T1 T2
Gene-Ontology Analysis on the Differentially Expressed Genes in Maize (Zea mays L.) Ear Rot 223
Fig. 3 Pathway graph of GO function analysis on cellular component. Pathway of the gene in the crosstalk was indicated in the cellular part.
Fig. 4 Pathway graph of GO function analysis on molecular function. Pathway of the gene participating in molecular function was indicated in the network.
against pathogen infection, such as MAPKs (mitogen-activated protein kinases). These differential
genes are important in the development, growth, and response to endogenous and environmental cues [20]. The group genes of molecular function were
associated with roles in plant defense activities in response to pathogen infection. Further analysis on the
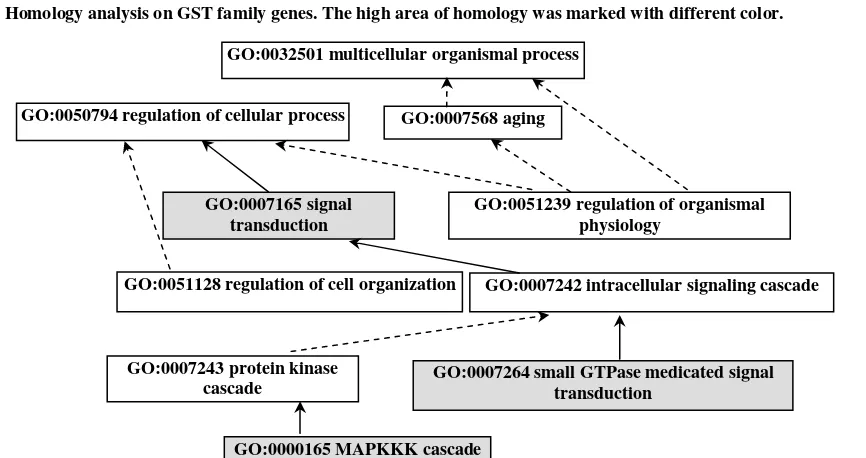
molecular function class by GO, a GST (glutathione-S-transferase) family with high homology was specifically elicited during molecular
annotation process (Fig. 5). As is known, the GST family is belonged to important antioxidant and
involved in catabolic process, which can be found widely in plant against pathogens. Of course, these
GST genes may play some other roles in physiological responses [21]. The present analysis could enhance our hypothesis that the accumulation of GST caused
by pathogens has been linked to reduce symptoms and higher levels of resistance after FM affection.
This information provided that GST genes may contribute to improve host tissues resistance to FM.
3.5 GO Annotation Involved in Biological Process
Differentially expressed genes in this class were
GO:0016722 transferase activity
GO:0016779 nucleotidetransferase activity
GO:0016301 kinase activity
GO:0016773 phosphotransferase activity
GO:0004306
ethanolamine-phosphate cytidyly
GO:0008905 mannose-phosphate guanylyltansferase
GO:0004475 mannose1-phosphate
guanylyltansferase
GO:0004674 protein serine/threonine
GO:0050321 tau-protein kinase
activity
GO:0004712 protein serine/threonine kinase
GO:0004708 MAP kinase kinase activity GO:0004672 protein
kinase activity GO:0016029
membrane
GO:0044425 membrane part
GO:0044459 plasma membrane part
GO:0005886 plasma membrane
GO:0031224 intrinsic to membrane
GO:0031226 intrinsic to membrane
GO:0005887 intergral to plasma membrane
GO:0031225 anchored to membrane
Gene-Ontology Analysis on the Differentially Expressed Genes in Maize (Zea mays L.) Ear Rot 224
Fig. 5 Homology analysis on GST family genes. The high area of homology was marked with different color.
Fig. 6 Pathway graph of GO function analysis on biological process. Pathway of the gene participating in biological process was indicated in the network.
associated with various biological process and
complicated network in cell, including protein synthesis and destination, transcriptional regulation, metabolism and energy, growth and development,
signal transduction pathway or defensive reaction. As showed in our analysis result, MAPK and CDPK
(calcium-dependent protein kinase) pathways were identified by GO elucidation (Fig. 6). The two signal transduction pathways were particularly presented
here, suggesting that this class genes associated with
complicated biological process could regulate plant defense responses to FM infection. Everyone knows that a pathogen-induced disease could induce multiple
cellular activities, including various physiological changes, membrane integrity, DNA-protein interaction,
and gene expression [22]. The striking finding in our analysis could provide important information that the signal transduction pathways may play an important
GO:0032501 multicellular organismal process
GO:0050794 regulation of cellular process GO:0007568 aging
GO:0007165 signal transduction
GO:0051239 regulation of organismal physiology
GO:0051128 regulation of cell organization GO:0007242 intracellular signaling cascade
GO:0007243 protein kinase cascade
GO:0007264 small GTPase medicated signal transduction
GO:0000165 MAPKKK cascade
Gene-Ontology Analysis on the Differentially Expressed Genes in Maize (Zea mays L.) Ear Rot 225
role in the host defense against FM. Moreover, genes involved in other biological process also could play
complicated roles in defense system.
4. Conclusion
Ear rot disease in maize has a direct effect on maize
kernels and bract tissues; thus, investigation of the defense responses that occur in bract tissues following inoculation with FM will improve our understanding
of the host–pathogen interaction. It widely known that massive information could be generated basing on
genechip application in the plant resistant genes screening. In our previous works, we have isolated and identified large number induced genes association
with resistance to Fusarium ear rot. Based on microarray platform, 482 genes were significantly
induced in Bt-1 after FM infection. The role of the FM-induced genes in the genechip data in conferring disease resistance requires further investigation, and
altering the levels of such genes might play critical roles in modulating or enhancing resistance to FM
affection. This GO analysis is a continuation of the previous report.
Bioinformatics analysis based on molecular
function annotation is an important research method in predicting gene function and has been applied in many
species for deeply digging further information [23]. In this study, it was attempted to analyze the detail physiological function of large number genes in the
process of defense system in maize ear rot by GO annotation. GO analysis performed here indicates that
the interaction between maize and FM results in a range of differentially expressed genes encoding related important proteins in plant defense, signal
transduction, and regulation of transcription, particularly in resistant inbred line Bt-1. All the
FM-induced genes were classified into cellular component, molecular function and biological process with complicated network by molecular annotation
system, which could provide information on physiological responses during FM inoculation. The
FM-induced genes associating with the three
classifications in Bt-1 might play important roles in modulating the response to FM infection or enhancing
plant protection system.
In conclusion, the GO analysis is the further step
for large number of genes toward better understanding of the molecular responses in maize ear rot. The presented results are a valuable guide for further
functional genomics studies addressing resistant mechanisms in maize ear rot. Further functional
analysis of these FM-induced genes to FM may provide new insights into the molecular mechanisms of the host defense response. In future studies, the
involvement of each gene in FM inducement should be investigated and will help us clarify the process of
defense mechanisms acquisition combating invasion in maize ear rot.
Acknowledgments
This research was supported by the Natural
National Science Foundation of China (No. 30571173, No. 31201274), National High Technology Research
and Development Program of China (863 Program) (No. 2012AA10A307).
References
[1] L.M. Reid, R.W. Nicol, T. Ouellet, Interaction of
Fusarium graminearum and F. moniliforme in maize ears: Disease progress, fungal biomass, and mycotoxin accumulation, Phytopathology 89 (1999) 1028-1037. [2] L.A. Robertson-Hoyt, M.P. Jines, P.J. Balint-Kurti, QTL
mapping for Fusarium ear rot and Fumonisin
contamination resistance in two maize populations, Crop Science 46 (2006) 1734-1743.
[3] M. Weidenborner, Foods and fumonisins, European Food Research and Technology 212 (2001) 262-273.
[4] C.J. Wen, X.J. Chen, W.R. Chen, Fusarium ear rot of corn and methods used in resistance test, Journal of Sichuan Agricultural University 20 (2002) 321-323. [5] M.L. Ali, J.H. Taylor, L. Jie, Molecular mapping of
QTLs for resistance to Gibberella ear rot, in corn, caused by Fusarium graminearum, Genome 48 (2005) 521-533. [6] G.P. Munkvold, Epidemiology of Fusarium diseases and
their mycotoxins in maize ears, European Journal of Plant Pathology 109 (2003) 705-711.
Gene-Ontology Analysis on the Differentially Expressed Genes in Maize (Zea mays L.) Ear Rot 226
Journal of Heredity 87 (1996) 382-385.
[8] B. Vigier, L.M. Reid, L.M. Dwyer, Maize resistance to gibberella ear rot: Symptoms, deoxynivalenol, and yield, Canadian Journal of Plant Pathology 23 (2001) 99-105. [9] M.C. Heath, Hypersensitive response-related death, Plant
Molecular Biology 44 (2000) 321-334.
[10] J.M. Casacuberta, D. Raventos, P. Puigdomenech, Expression of the gene encoding the PR-like protein PRms in germinating maize embryos, Molecular Genetics and Genomics 234 (1992) 97-104.
[11] M.J. Cordero, D. Raventos, B.S. Segundo, Expression of a maize proteinase inhibitor gene is induced in response to wounding and fungal infection: Systemic wound response of a monocot gene, Plant Journal 6 (1994) 141-150. [12] I. Murillo, E. Jaeck, M.J. Cordero, Transcriptional
activation of a maize calcium-dependent protein kinase gene in response to fungal elicitors and infection, Plant Molecular Biology 45 (2001) 145-158.
[13] D.S. Multani, R.B. Meeley, A.H. Paterson, Plant-pathogen microevolution: Molecular basis for the origin of a fungal disease in maize, PNAS 95 (1998) 1686-1691.
[14] G.S Johal, S.P. Briggs, Reductase activity encoded by the HM1 disease resistance gene in maize, Science 258 (1992) 985-987.
[15] J. Yuan, M.L. Ali, J. Taylor, A guanylyl cyclase-like gene is associated with Gibberella ear rot resistance in
maize (Zea mays L.), Theoretical Applied Genetics 116 (2008) 465-479.
[16] F. Zhang, X.Q. Wan, G.T. Pan, QTL mapping of
Fusarium moniliforme ear rot resistance in maize 1 map construction with microsatellite and AFLP markers, Journal of Applied Genetics 47 (2006) 9-15.
[17] C. Li, W.H. Wong, Model-based analysis of oligonucleotide arrays: Expression index computation and outlier detection, PNAS 98 (2001) 31-36.
[18] G.S. Yuan, Z.M. Zhang, K. Xiang, Large-scale identification of differentially expressed genes in maize inbreds susceptible and resistant to Fusarium ear rot, Plant Omics Journal 5 (2012) 471-475.
[19] P.M. Schenk, K. Kazan, J.M. Manners, Coordinated plant defense responses in Arabidopsis revealed by microarray analysis, PNAS 97 (2000) 11655-11660.
[20] S. Zhang, D.F. Klessig, MAPK cascades in plant defense signaling, Trends in Plant Science 6 (2001) 520-527. [21] E. Lebel, P. Heifetz, L. Thorne, Functional analysis of
regulatory sequences controlling PR-1 gene expression in Arabidopsis, Plant Journal 16 (1998) 223-233.
[22] T. Eulgem, Regulation of the Arabidopsis defense transcriptome, Trends in Plant Science 10 (2005) 71-78. [23] G.S. Song, H.L. Zhai, Y.G. Peng, Comparative
Mar. 2013, Vol. 7, No. 3, pp. 227-235
Journal of Life Sciences, ISSN 1934-7391, USA
Analysis of Methylation-Sensitive Amplified
Polymorphism and Prediction of Candidate
Genes Infected by SCMV in
Zea mays
Genome
Li Liu1, 2, Xiu-Jing He2, Zhi-Ming Zhang2, Mao-Jun Zhao3 and Guang-Tang Pan2
1 Personnel Department, Sichuan Agricultural University, Ya’an 625014, China
2 Maize Research Institute, Sichuan Agricultural University/Key Laboratory of Crop Genetic Resources and Improvement of Chinese Ministry of Education, Chengdu 611130, China
3 Department of Life Sciences, Sichuan Agricultural University, Ya’an 625014, China
Received: January 28, 2013 / Accepted: March 11, 2013 / Published: March 30, 2013.
Abstract: DNA methylation is an important component of the epigenetic network, and it plays important roles in gene expression regulation and epigenetic change response to various stresses. In this study, the authors assessed the methylation patterns stressed by SCMV (sugarcane mosaic virus) in maize by methylation-sensitive amplified polymorphism (MSAP), and identified important candidate genes related to SCMV resistance through combining microarray analysis with CpG islands prediction. The results of MSAP indicated DNA methylation levels appeared dynamic changes inoculated for 0 d, 1 d, 4 d, 5 d and 10 d. 118 candidate genes were identified infected by SCMV, which may participate in DNA methylation modification. Among them, eight candidate genes were mapped on Scmv1 and Scmv2 QTL regions, which are crucial for SCMV resistance. In conclusion, DNA methylation is closely related with maize resistance to SCMV and plays an important role in regulating gene expression responded to maize resistance.
Key words: Maize, SCMV, DNA methylation, MSAP, CpG islands.
1. Introduction
Biotic and abiotic stresses are important limiting factors for crop growth and improvements in yields. Moreover, they are huge evolutionary forces causing genetics mutation and epigenetic changes. In eukaryotic genomes, epigenetics provide stability and diversity to the phenotype through epigenetic modifications that affect local transcriptional potential and that are preserved or regenerated during organisms growth and development. Epigenetic modifications consist mainly of DNA methylation, histone modifications, nucleosome location and expression of non-coding RNA [1-3]. Plants employ epigenetic regulatory strategies, such as DNA
Corresponding author: Guang-Tang Pan, Ph.D., professor, research fields: crop genetics and breeding. E-mail: [email protected].
methylation, to maintain this genomic plasticity, allowing relatively rapid adaptation to new conditions and regulating genome functions to a large extent without changing the DNA sequence. Significant differences in DNA methylation levels, especially methylation of cytosine, have been observed among various tissue types and different growth stage in higher plants, which contribute to the control of gene expression to response to stress.
Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction
of Candidate Genes Infected by SCMV in Zea mays Genome
228
is found in vertebrates, plants and fungi [5]. CpGs are enriched in short stretches of CpG-dense DNA known as CpG islands. CpG islands often appear to be frequent targets of hypermethylation events [6, 7], so they are treated as the focus in DNA methylation research.
A series of developments in the methods were used to detect DNA methylation. Recently developed methods can be classified into three main approaches according to the principles used in detection: endonuclease digestion, affinity enrichment and bisulphite conversion [3]. Methylation-sensitive amplified polymorphism (MSAP) analysis is based on endonuclease digestion, and use one pair isoschizomers for detection of DNA methylation. It is a modification of amplified fragment length polymorphism (AFLP) technique, in which the isoschizomers HpaII and MspI are employed as “frequent-cutter” enzymes, to assess the extents and patterns of cytosine methylation [8]. Moreover, MSAP have become an important tool for characterization of DNA methylation in heterosis analysis, developmental regulation and stress responses [9-12].
The SCMV (sugarcane mosaic virus) is major pathogens of maize worldwide. It is one of the most important virus diseases of maize, resulting in significant yield losses and economic losses in maize. Scmv1 and Scmv2 were mapped to chromosome arms 6S and 3L, have been shown to confer complete resistance to SCMV [13, 14]. However, the molecular mechanisms underlying resistance to SCMV in maize have not been extensively characterized. Nowadays, a large number of maize microarray experiments are accessible via different public resources such as arrayexpress or GEO, and offer the chance to identify and characterize important gene relate to resistance and get a deep insight into DNA methylation mechanism of biotic stress in maize. In this study, the authors assess the extents and patterns of DNA methylation by MSAP in maize, identify candidate genes associated with DNA methylation changes using microarray and propose the molecular
mechanisms underlying the development and progression of SCMV infection.
2. Materials and Methods
2.1 Plant Material and Treatments
In the present study, inbred lines Huangzao 4 (highly resistant to SCMV) and Mo17 (highly susceptible to SCMV) were grown and maintained under controlled greenhouse conditions. The sap for the inoculation was produced by homogenizing the infected leaves in 0.05 M potassium phosphate buffer, pH 7.2 (1:10, w/v). Plants at the 3- to 4-leaf stage were used for virus inoculations using mechanical rub inoculation [15]. Non-infected plants and infected plants were kept in separate growth chambers after inoculation. Infected leaves were collected at five time points (0 d, 1 d, 4 d, 5 d and 10 d) after inoculation and immediately frozen in liquid nitrogen and total genomic DNA extracted following the CTAB method [16].
2.2 Methylation-Sensitive Amplification Polymorphism Analysis
To detect MSAP, restriction and ligation were done concurrently and two consecutive PCRs, including pre-selective amplification and selective amplification, were used to selectively amplify the EcoRI–HpaII and EcoRI–MspI DNA fragments. The adapters and primers for enzyme “rare- cutter” EcoRI was the same as that used in standard AFLP analysis, while enzyme “frequent-cutter” HpaII/MspI adapter was designed according to Xiong et al. [17]. MSAP analysis was carried out according an established protocol [17]. The second PCR products were separated by electrophoresis on 6% sequencing gels and stained with silver as described by Bassam et al. [18].
2.3 CpG Island Predition and Candidate Gene Identification
Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction
of Candidate Genes Infected by SCMV in Zea mays Genome
229
gene expression atlas (E-MEXP-253 [19], E-TABM-586 [15]) of the maize under SCMV stress were downloaded from the publicly available databases arrayexpress (http://www.ebi.ac.uk/ arrayexpress/). Microarray data were analysed using the TIGR Microarray Data Analysis System as described by Shi et al. [19] and Anna Użarowska et al. [15]. Differentially expressed ESTs were used to query B73 genome sequences (http:// www.maizesequence.org/index.html) using blast tool. Parameters were as followings: maximum identity > 95%, length > 200 bp and E value < 1010.
To investigate DNA methylation in promoter regions, 3 kb of genomic DNA sequences upstream of initiation codon ATG were retrieved from the B73 maize sequencing database and searched against the
PLACE database (http:// www.dna.affrc.go.jp/PLACE/). Subsequently, differentially expressed genes sequences and promoter
regions were used to predict CpG islands by CpGPAP [20]. Parameters were as followings: CpG minimum length exceeds 200 bp, the observed/expected (O/E) ratio surpasses 0.65 and minimum GC content is greater than 55%, other parameters took default values.
GO annotation of the candidate genes were analysed by Goanna tools in AgBase (http://www.agbase.msstate.edu), and plotted by WEGO [21]. Candidate genes genetic map positions were determined using the MaizeGDB (http://www.maizegdb.org/).
3. Results
3.1 Plants Phenotype Analysis after Inoculation with
SCMV
With the increase stress time, the mosaic symptoms became more and more serious in susceptible Mo17. Typical mosaic symptoms, mosaic and chlorosis, were observed in leaves of susceptible Mo17 10 d after inoculation with SCMV, whereas Huangzao4 displayed no SCMV symptoms.
3.2 Methylation Profiles in Inoculated Plants
Comparison of the fragments obtained after digestions with EcoRI/HpaII and EcoRI/MspI of DNA from maize at five time points after inoculation, revealed four main kinds of patterns: (1) The 5′-CCGG-3′ is unmethylated or hemi-methylated (single strand) at the internal cytosine; both HpaII and MspI recognize the site and cut the DNA, product the same fragments in HpaII and MspI electrophoresis lanes. (2) The 5′-CCGG-3′ is hemi-methylated at the external cytosine or at both cytosines lead to the appearance of a fragment generated from the EcoRI/HpaII digest, but not in that obtained from the EcoRI/MspI digest. (3) The HpaII/MspI recognition site is fully methylated (both strands) at the internal cytosine; MspI cuts, whereas HpaII does not. (4) Furthermore, make two isoschizomers are insensitive to fully methylation occur at external or both external and internal cytosines, no fragments are produced with either HpaII or MspI [22].
144 pairs of primers were detected in pre-selective amplification, in order to determine conditions that would yield distinct amplified fragments on the sequencing gel. Among them, 20 pairs of primers were screened and used to detect cytosine methylation at the 5′-CCGG-3′ sequence in Huangzao4. A total of 3,712 fragments in 688 fragment sites resolved by 20 primer pair combinations were detected by MSAP in DNA extracted from various time-points (Table 1). For each primer combination, each individual time-point displayed approximately 35 fragment sites, each of the fragment sites represented a recognition site cleaved by one or both of the isoschizomers. Of the 3,712 fragments, 788 were differentially amplified from the two digests for at least one of five time points, due to differentially sensitivity of HpaII and MspI to cytosine methylation.
Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction
of Candidate Genes Infected by SCMV in Zea mays Genome
230
Table 1 Methylation levels of 5 time-point after inoculation in maize.
Time-point
Pattern
Total sites Ⅰ Ⅱ Hemi-methylation
ratio (%)a Ⅲ IV Ⅲ+IV
Fully methylation ratio (%)b
Methylation ratio (%)c
0 d
688
283 77 11.19 80 248 328 47.67 58.86
1 d 302 76 11.04 73 237 310 45.05 56.09
4 d 297 80 11.62 68 243 311 45.2 56.82
5 d 307 66 9.59 67 248 315 45.78 55.37
10 d 273 99 14.38 102 214 316 45.93 60.31
a: Hemi-methylation ratio = II/(I + II + III + IV), b: Fully methylation ratio = (III+IV)/(I+II+III+IV), c: Methylation ratio = (II + III + IV)/(I + II + III + IV).
maize plants, unmethylation sites, hemi-methylation sites, and fully methylation sites equal to 41.13%, 11.19% and 47.67% of 688 fragments sites. In detail, hemi-methylation sites accounted for 19.01% of the total methylation ratio (approximately 58%), the remaining percentages were ascribed to fully methylation sites (Table 1). Huangzao4 showed a significant hypermethylation in the 5′-CCGG-3′ sequence similar to genome-wide methylation of B73 inbred lines [23]. This high level of DNA methylation in 5′-CCGG-3′ sequence was consistent with hypermethylation of cytosine [24, 25].
3.3 Changes in Patterns of Cytosine Methylation between Five Time-points Revealed by MSAP
To seek to clarify the relative levels of DNA methylation variation among different time-points in maize under SCMV stress, the authors assessed changes in patterns of cytosine methylation between five time-points, as detected by MSAP. These results showed that the general trend of the methylation ratio descreased with time-course had only risen slightly at 10 d after inoculation. Hemi-methylation ratio and fully methylation ratio presented similar trends (Table 1). The situation showed a reverse trend to accumulation of HC-Pro, which is a helper component proteinase produced from SCMV (Table 1) [26]. In the early stages of infection, expression levels of HC-Pro increased and reached the maximum 9 d after inoculation, subsequently showed gradually trending down. On the contrary, the levels of cytosine
methylation increased after an obvious decline. In combination, these results suggested that maize regulate gene expression to resist SCMV by changing DNA methylation patterns.
In detail, changes in methylation patterns among four time-points (0 d, 1 d, 4 d and 5 d) were analysed by comparing patterns of amplified fragments in single sites. Three major classes of changes patterns were identified among the differentially amplified fragments (Table 2). In the first class, the same methylation sites were detected in all time-points; these are referred to as monomorphic with respect to cytosine methylation. There were 278 sites detected by 20 primer pairs presented monomorphic, 131 sites reflected unmethylation in cytosine, and 147 fragments were the results of hemi-methylation and fully methylation.
Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction
of Candidate Genes Infected by SCMV in Zea mays Genome
231
Table 2 Changes of cytosine methylation between 0 d time-point and 5 d time-point in the Huangzao4.
changes in patterns 0 d 5 d Number of sites Ratio
Not polymorphic Ⅰ Ⅰ 131 19.04%
Ⅱ Ⅱ 9 1.31%
Ⅲ Ⅲ 23 3.34%
IV IV 74 10.76%
IV Ⅲ 13 1.89%
Ⅲ IV 28 4.07%
Demethylation Ⅱ Ⅰ 24 3.49%
Ⅲ Ⅰ 28 4.07%
Ⅲ Ⅱ 3 0.44%
IV Ⅰ 124 18.02%
IV Ⅱ 35 5.09%
Methylation Ⅰ Ⅱ 21 3.05%
Ⅰ Ⅲ 30 4.36%
Ⅰ IV 99 14.39%
Ⅱ Ⅲ 2 0.29%
Ⅱ IV 44 6.40%
Total 688 100.00%
methylation polymorphism with 23.11% and 20.79% sites, respectively. On the whole, the demethylation sites were higher methylation sites, resulted in decline of methylation in cytosine.
It is worth noting that 26% (data unshown) sites recovered to previous metylation patterns at 10 d after in inoculation with SCMV. These sites may be not involved in the Huang4 response to SCMV stress, but resulted from the secondary effects produced by changes of the methylation status. In addition, these sites partly reflected high resistance of Huangzao4. The specific functions of these sites need further investigation.
3.4 Candidate Genes Identification and CpG Island Predition
CpG islands are DNA regions that contain a high frequency of CpG dinucleotides relative to their occurrence in the bulk genome, often appear to be frequent targets of hypermethylation events [6, 7]. So, the authors extracted differential expression candidate genes under SCMV stress from microarray data, and then identified important candidate genes by CpG
islands prediction. These important candidate genes had higher probability of DNA methylation changes in comparison with the others, and may be regulated by DNA methylation changes in expression levels.
Microarray-based expression analysis has revealed that 118 differential expression candidate genes may participate in responses to SCMV stress, among which 77 genes were up-regulated and 41 genes were down-regulated under SCMV stress (data unshown). Furthermore, CpG Islands characteristics of candidate genes were analysed using CpGPAP [20]. In addition, promoter regions of candidate genes, obtained from B73 maize sequencing database, were predicted through the same method.
Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction
of Candidate Genes Infected by SCMV in Zea mays Genome
232
them, eight important candidate genes were located in Scmv1 and Scmv2 regions (Fig. 2a). As shown in Fig. 2b, the results of cis-acting regulatory elements in promoter showed that CGCG-BOX was major elements, which involved in signal transduction pathway and regulation of plant hormone. W-BOX, EBOX, MYC recognition site also were found in promoter regions of candidate genes, which were transcription factor binding site. They are involved in many biological processes, such as plant development, metabolism, and responses to biotic and abiotic stresses.
3.5 GO Classification of Candidate Genes Affected by DNA Methylaiton Modification
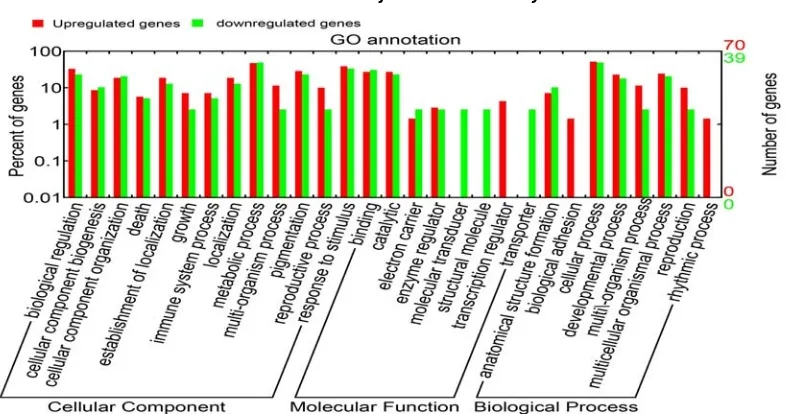
Go annotation of differentially expressed genes association with SCMV stress found that the differentially expressed genes involved in biological process such as response to stimulus, signal transmission and molecular function such as cabalistic and transcription regulation (Fig. 3). Diverse categories were observed, it is interesting that the numbers of genes in molecular function are significantly different between up- and down-regulation (Fig. 3). This result reveals that maize predominantly depend on up- regulated transcription regulator for response to SCMV stress.
4. Discussion
4. 1 Biotic Stress-Induced Methylation Alteration
Several environmental and genetic stimuli are known to induce DNA methylation alteration in plants. 5mC has been described as an obligatory component of transcriptionally silent chromatin, and was considered widely to be a mechanism that protected the genome against transposable elements and retro-viruses, and suppressed the activity of repetitive sequences and pseudogenes [6, 27]. In the present study, MSAP approach was used to confirm whether SCMV stress caused DNA methylation changes and assess the extents and patterns of cytosine methylation in maize.
The results showed the dynamic changes of methylation in five time-points after inoculation with SCMV. The opposite trends between DNA methylation and HC-Pro revealed DNA methylation played an important role in stress responses. Changes in DNA methylation can be considered either passive reactive of SCMV stress or active defensive mechanism for regulating the gene expression. In this study, some sites, recovered to previous metylation patterns, may be involved in passive reactive of SCMV stress. Another site maybe regulate the gene expression to resist SCMV through active defensive mechanism.
(A) (B)
Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction
of Candidate Genes Infected by SCMV in Zea mays Genome
233
Fig. 3 Gene ontology annotation of candidate genes. Red bars indicated up-regulated genes and green bars represented down-regulated genes.
Establishing, maintaining and modifying DNA methylation patterns are critical for diverse biological processes. In plants, de novo methylation is catalyzed by Domains rearranged methyltransferase 2 (DRM2) and maintained by DNA methyltransferase 1 (MET1), Chromomethylase (CMT3) and DRM2 in CpG, CpNpG and CpNpN methylation, respectively [28]. A number of studies have revealed the involvement of small interfering RNAs (siRNAs) in RNA-directed DNA methylation (RdDM), and small RNAs seem to be important factors to determine the distribution of chromatin modifications [29, 30]. Although in most cases DNA methylation is a stable, reduced levels of methylation are observed in plants under stress. Demethylations were processed by enzyme reaction and DNA replication [31]. However, DNA is scarcely replicated in stressed tissues, so demethylation must be processed by enzyme reaction mechanism. It is worth noting that demethylations were higher than methylation during SCMV infection, which leaded to up-regulated expression of gene related to resistance. From the mentioned above, the analysis of MSAP suggested that SCMV stress induce DNA methylation changes, which may be a key defence mechanism in response to SCMV stress in maize.
The authors have adapted MSAP technique for detection of cytosine methylation in the maize genome
under SCMV stress. The results showed that this technique is highly efficient for large-scale detection of cytosine methylation in the maize genome. However, it should be pointed out, this method can only investigate a part of the cytosines in the genome, leading to cannot fully reflect levels of DNA methylation. Another constraint ascribed to isoschizomers, the isoschizomers did not allow us to distinguish unmethylation from hemi-methylated at the internal cytosine.
4.2 Important Candidate Genes Affected by DNA Methylation
Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction
of Candidate Genes Infected by SCMV in Zea mays Genome
234
resistance in earlier stages of plant development, whereas Scmv2 at later stages of infection [12, 13, 33]. Another candidate gene is localized in other genome regions may act further downstream in the signal transduction pathway or induced by resistance genes located in the Scmv1 and Scmv2 regions.
These candidate genes affected by DNA methylation contained multiple cis-regulatory elements, such as CGCG-BOX, W-BOX, E-BOX and MYC recognition site, which were recognized by transcription factor, involved in plant development, metabolism, signal transduction and responses to biotic and abiotic stresses. The authors identified these important candidate genes will be useful for further investigating DNA methylation changes under SCMV stress conditions.
4.3 CpG Islands and Regulation of Transcription
DNA methylation has temporal and spatial specific, and have been proposed to play a key role in epigenetic variation that could provide adaptation to environmental stresses. In plants, DNA methylation major involved in the control of all genetic functions including transcription, replication, DNA repair, gene transposition and cell differentiation. In addition, DNA methylation is engaged in gene silencing and parental imprinting, and controls expression of transgenes and foreign DNA in cell. Studies showed that a large class of CpG islands is remote from annotated transcription start sites (TSSs), and unmethylated CpG islands are organized in a characteristic chromatin structure that predisposes them toward promoter activity. These findings emphasize the strong correlation between CpG islands and transcription initiation [34-36]. GC richness increases the probability that ubiquitous transcription factors will bind [37]. The data of cis-regulatory elements and CpG islands prediction in promoter showed that there are many cis-regulatory elements co-localized with CpG islands, the banding between transcription factors and cis-regulatory elements may be regulated by DNA methylation modification in
CpG islands regions.
5. Conclusion
DNA methylation is a fundamental mechanism of epigenetic modification, which often carries on bidirectional adjustment to regular genes expression. It is also a defense mechanism to response to stress. Heavy methylation of cytosine residues plays an important role in gene expression, and the MSAP technique may provide a very useful tool for comparative assessment of the patterns and extents of DNA methylation. In the present study, the authors assessed DNA methylation changes under SCMV stress by MSAP, and identified a total of 118 candidate genes related to resistance. Among them, eight candidate genes were mapped on Scmv1 and Scmv2 QTL regions, which are crucial for complete resistance to SCMV. These studies will be useful for expounding an important role for DNA methylation in response to biotic stress.
References
[1] G. Riddihough, L.M. Zahn, What is epitigenecs?, Science 330 (2010) 611.
[2] S. Feng, S.E. Jacobsen, W. Reik, Epigenetic reprogramming in plant and animal development, Science 330 (2010) 622-627.
[3] P. W. Laird, Principles and challenges of genome-wide DNA methylation analysis, Nature Genetics 11 (2010) 191-203.
[4] C.S. Pikaard, Nucleolar dominance and silencing of transcription, Trends Plant Sci 4 (12) (1999) 478-483. [5] A. Kovarik, R. Matyasek, A. Leitch, B. Gazdova, J
Fulnecek, M. Bezdek, Variability in CpNpG methylation in higher plant genomes, Gene 204 (1-2) (1997) 25-33. [6] A. Bird, DNA methylation patterns and epigenetic
memory, Genes 16 (2002) 6-21.
[7] K.D. Robertson, P.A. Jones, DNA methlation: Past, present and future directions, Carcinogenesis 21 (2000) 461-467.
[8] M.F. Fraga, M. Esteller, DNA Methylation: A profile of methods and applications, Biotechniques 33 (2002) 632-649.
Analysis of Methylation-Sensitive Amplified Polymorphism and Prediction
of Candidate Genes Infected by SCMV in Zea mays Genome
235
[10] R. Alina, S. Sgorbati, A. Santagostino, M. Labra, A. Ghiani, M. Citterio, Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp, Physiol. Plant 121 (2004) 472-480.
[11] I. Ashikawa, Surveying CpG methylation at 5’-CCGG in the genomes of rice cultivars, Plant Mol. Biol. 45 (1) (2001) 31-39.
[12] Z. Yi, Y. Sun, T. Niu, X. Liang, L. Liu, M. Yan, et al., Genomic DNA cystoine methylation of corn hybrids and their parents, Acta Botanica Boreali-Occidenaliat Sinica 25 (12) (2005) 2420-2425.
[13] X. Xia, A.E. Melchinger, L. Kuntze, T. Lübberstedt, Quantitative trait loci mapping of resistance to sugarcane mosaic virus in maize, Phytopathology 89 (8) (1999) 660-667.
[14] C.M. Dussle, A.E. Melchinger, L. Kuntze, A .Stork, T. Lübberstedt, Molecular mapping and gene action of Scm1 and Scm2, two major QTL contributing to SCMV resistance in maize, Plant Breed 119 (4) (2000) 299-303. [15] A. Użarowska, G. Dionisio, B. Sarholz, H.P. Piepho, M.
Xu, C. RønnIngvardsen, et al., Validation of candidate genes putatively associated with resistance to SCMV and MDMV in maize (Zea mays L) by expression profiling, BMC Plant Biology 9 (2009) 15.
[16] Doyle JJ, Doyle JL, Isolation of plant DNA from fresh tissue, Focus 12 (1990) 13-14.
[17] L. Xiong, C. Xu, S. Maroof, Q. Zhang Q, Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique, Mol. Gen. Genet. 261 (1999) 439-446.
[18] B.J. Bassam, G. Caetano-Anolles, P.M. Gresshoff, Fast and sensitive silver staining of DNA in polyacrylamide gels, Anal. Biochem. 19 (1991) 680-683.
[19] C. Shi, F. Thümmler, A.E. Melchinger, G. Wenzel, T. Lübberstedt, Comparison of transcript profiles between near-isogenic maize lines in association with SCMV resistance based on unigene-microarrays, Plant Science 170 (2006) 159-169.
[20] L. Chuang, C. Yang, M. Lin, C. Yang, CpGPAP: CpG island predictor analysis platform, BMC Genetics 13 (2012) 13.
[21] J. Ye, L, J. C. Fang, H. Zheng, Y. Zhanghen, Z. Zhang, et al., WEGO: A web tool for plotting GO annotations, Nucleic Acids Research 34 (2006) 293-297.
[22] A. Sha, H. Lin, J. Huang, D. Zhang, Analysis of DNA methylation related to rice adult plant resistance to bacterial blight based on methylation-sensitive AFLP, Mol Genet Genomics 273 (2005) 484-490.
[23] S.R. Eichten, R.A. Swanson-Wagner, J.C. Schnable, A.J.
Waters, P.J. Hermanson, S. Liu, et al., Heritable epigenetic variation among maize inbreds, PLoS Genet 7 (11) (2011) e1002372.
[24] A.M. Deaton, A. Bird, CpG islands and the regulation of transcription, Genes 25 (2011) 1010-1022.
[25] A.P. Bird, CpG-rich islands and the function of DNA methylation, Nature 321 (1986) 209-213.
[26] Z. Xia, P. Zhou, K. Wu, J. Wu, Cloning and Expression Analysis of the HC-Pro Gene from Isolate of Sugarcane Mosaic Virus Infecting Maize, Journal of MaizeSciences 18 (3) (2010) 155-159.
[27] R.A. Martienssen, V. Colot, DNA methylation and epigenetic inheritance in plants and filamentous fungi, Science 293 (2001) 1070-1074.
[28] J.A. Law, S.E. Jacobsen, Establishing, maintaining and modifying DNA methylation patterns in plants and animals, NatRev Genet 11 (3) (2010) 204-220.
[29] V. Chinnusamy, J. Zhu, RNA-directed DNA methylation and demethylation in plants, Sci. China C. Life Sci. 52 (4) (2009) 331-343.
[30] X. He, T. Chen, J. Zhu, Regulation and function of DNA methylation in plants and animals, Cell Research 21 (2011) 442-465.
[31] J.P. Jost, E.J. Oakeley, B. Zhu, D. Benjamin, S. Thiry, M. Siegmann, et al., 5-Methylcytosine DNA glycosylase participates in the genome-wide loss of DNA methylation occurring during mouse myoblast differentiation, Nucl. Acids Res. 29 (21) (2001) 4452-4461.
[32] L. Yuan, C.M. Dussle, A.E. Melchinger, H.F. Utz, T. Lübberstedt, Clustering of QTL conferring SCMV resistance in maize, Maydica 48 (2003) 55-62.
[33] M. Xu, A.E. Melchinger, X. Xia, T. Lübberstedt, High-resolution mapping of loci conferring resistance to sugarcanemosaic virus in maize using RFLP, SSR, and AFLP markers, Mol. Gen. Genet 261 (1999) 574-581. [34] S. Saxonov, P. Berg, D.L. Brutlag, A genome-wide
analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters Proc. Natl. Acad. Sci. USA 103 (5) (2006) 1412-1417.
[35] P. Carninci, A. Sandelin, B. Lenhard, S. Katayama, K. Shimokawa, J. Ponjavic et al, Genome-wide analysis of mammalian promoter architecture and evolution, Nat. Genet. 38 (2006) 626-635.
[36] T. Juven-Gershon, J.Y. Hsu, J.W. Theisen, J.T. Kadonaga, The RNA polymerase II core promoterthe gateway to transcription, Curr. Opin. Cell Biol. 20 (2008) 253-259. [37] R.S. Illingworth, U. Gruenewald-Schneider, S. Webb,
Mar. 2013, Vol. 7, No. 3, pp. 236-243
Journal of Life Sciences, ISSN 1934-7391, USA
BRCA1 Mutation Detection Using Fluorescent
Hybridization Probes and Melting Curves
Safa R. Fitouri1, Nouri B. Ermeli2, Salah M. Bensaber1, 3, Mousa I. Jaeda3, Ibrahim A. Mrema3, Anton Hermann4 and Abdul M. Gbaj1, 3
1. Department of Genetics, National Medical Research Centre, Zawia, Z16, Libya
2. Department of Natural Products, Faculty of Pharmacy, University of Tripoli, Tripoli, M16, Libya 3. Department of Medicinal Chemistry, Faculty of Pharmacy, University of Tripoli, Tripoli, M16, Libya
4. Department of Cell Biology, Division of Cellular and Molecular Neurobiology, University of Salzburg, Salzburg A-5020, Austria
Received: November 09, 2012 / Accepted: January 05, 2013 / Published: March 30, 2013.
Abstract: The BRCA1 (Breast Cancer Anti-estrogen resistance-1), early-onset gene is expressed in cells of breast and other tissue and helps to repair damaged DNA or destroy cells in cases DNA cannot be repaired. When the BRCA1 gene is damaged, then the DNA is not repaired appropriately and this enhances the risk for cancer. Fluorescence and UV-visible thermal studies were performed for WT (wild type) and MT (mutant type targets) full systems. The target DNAs used were in the form of short oligonucleotides, genomic DNA. The probe system was used for detection of WT and SNP alleles of human BRCA1 [(170-190, G→T) and (290-310, G→T)]. The Cy5 dye attached to a probe oligonucleotide (10-mer) undergoes a fluorescence intensity change on hybridisation of the probe to the WT compared to MT targets. Our results indicate that the system consisting of the target sequence and the one probe oligonucleotides bearing the Cy5 dye assemble correctly at the specified target. Once the full system (probe and target) is arranged under suitable conditions, a red-shift emission and change in fluorescence intensity are seen at a suitable wavelength. Thermal studies also showed significant differences in Tmbetween WT and MT. The results suggest that the differences in the fluorescence intensity at 665 nm and the spectrophotometric Tm(s) for the WT and MT can be attributed to the type of binding of the probe to the target. The systems were sensitive to single nucleotide polymorphisms and this may help in high throughput applications in genetic testing and molecular diagnostics.
Key words: DNA, fluorescence probe, BRCA1 gene, melting temperature, anisotropy.
1. Introduction
Many genetic diseases have been found to be the result of a change of a single base pair. These alterations, termed SNP (single nucleotide polymorphisms), may cause changes in the amino acid sequence of important proteins [1, 2]. Some SNPs on the other hand may not cause a change in protein expression, but may be close on the chromosome to other unknown deleterious mutations, and can thus serve as genetic markers. In order to detect specific
Corresponding author: Abdul M. Gbaj, Ph.D., assistant professor, research fields: genetics and biochemistry. E-mail: [email protected].
BRCA1 Mutation Detection Using Fluorescent Hybridization Probes and Melting Curves 237
been chosen, an appropriate label must be applied, so that hybridisation to the target can be detected [4].
Breast cancer is the most common carcinoma in women. The increasing risk for the disease is 10% up to the age of 80 years. A family history of breast cancer and ovarian cancer is an important risk factor. Some 5%-10% of all cases of breast cancer and 25%-40% of cases in patients under the age of 35 years have a hereditary origin. BRCA1/BRCA2 mutations are responsible for 3%-8% of all cases of breast cancer [5, 6]. One million females worldwide are diagnosed with breast cancer every year. Treatment of advanced breast cancer is useless, so the early diagnosis has a high precedence in medical treatment of the disease [7]. The risk factors of breast carcinoma could be genetic, environmental or hormonal [5, 6].
Genetic risk factors play role in about 5%-10% of all cases, 90%-95% of them result from genetic mutation and approximately 5%-10% are inherited as dominant breast carcinoma susceptibility genes [5, 8]. Other genes were found to increase vulnerability to cancer and are also associated with familial breast cancer. These genes are BRCA1 (Breast Cancer Anti-estrogen resistance-1), BRCA2 (Breast Cancer Anti-estrogen resistance-2), ATM (Ataxia Telangiectasia Mutant gene), PTEN (Phosphate and Tensin homology) and P53 (Tumor Protein 53) [8]. There are also other less affecting genes involved with little importance [5, 8].
This study concentrates mainly on BRCA1 gene, a human tumor suppressor gene which produces a protein called breast cancer type 1 susceptibility protein. It originates in cells of the breast and other tissues such as the ovaries, where it helps to repair damaged DNA and obliterates cells if DNA cannot be repaired. If BRCA1 itself is damaged, the cells will be able to grow and eventually become carcinogen. The protein encoded by the BRCA1 gene as well other tumor suppressors, DNA damage sensors and signal transducers form a large multi-subunit protein
complex known as the BASC (BRCA1-associated genome surveillance complex) [9-11]. The human BRCA1 gene is located on the long (q) arm of chromosome 17 at band 21, from base pair 38,449,840 to base pair 38,530,994 (map). BRCA1 orthologs have been identified in most mammals for which complete genome data are available [11, 12]. In addition, the BRCA1 protein (known as RING finger protein 53) contains two zinc finger domains, the C3HC4 type (RING finger) and BRCA1 C terminus (BRCT) domain [13].
Some fluorescent dyes (e.g. the cyanine dyes Cy3 and Cy5) undergo changes in their fluorescence spectrum due to changes in their environment. Thus, the spectrum of such molecules attached to a probe oligonucleotide may change on hybridisation of the probe with the target and thus could be used as hybridisation probes [14, 15].
The aim of this study was to develop a system that can be used to detect specific target oligonucleotide sequences. The model system used comprised a target sequence plus one probe oligonucleotide bearing a Cy5 fluorophore. The probes will be Watson-Crick complementary to the target sequence. The probe will assemble at the target thereby bringing the Cy5 dye close to the DNA bases.
2. Material and Methods
Reagents of the highest quality available were purchased from the suppliers indicated. All water used was distilled and purified by ion exchange and charcoal using a MilliQ system (Millipore Ltd, UK). Tris buffer, PBS buffer and TB-buffer etc. were prepared from analytical reagent grade materials according to standard procedures. Values of pH were measured using a Hanna-Instruments HI 9321 microprocessor pH meter, calibrated with standard buffers (Sigma, UK) at 20 C.
BRCA1 Mutation Detection Using Fluorescent Hybridization Probes and Melting Curves 238
Oncology Institute, Libya. DNA was extracted from peripheral blood lymphocytes as indicated in Ref. [16]. Sample purity was checked by agarose gel electrophoresis and the concentration was estimated spectrophotometrically at A260 nm [16]. The ratio OD260/OD280 provides an estimate of the purity of the nucleic acid. Pure preparations of DNA have OD260/OD280 values of 1.8 to 2.0. If there is contamination with protein or phenol, the OD260/OD280 will be significantly less than the values given above. In these cases an accurate quantitation of the amount of nucleic acid will not be possible. DNA samples that deemed not to be sufficiently pure were subjected to re-purification by phenol/chloroform extraction technique.
DNAs (synthetic and genomic) were analysed using 3% agarose gels, which were made by heating a mixture of 1×TAE (Tris-Acetate-EDTA) buffer (50 mL), agarose (1.5 g) and an aliquot (5 µL) of Cyper Gold (1:10000 in DMSO). After cooling the gels, they were cast and loaded in a plastic gel box covered with 1×TAE buffer (200 mL) [16]. Once the gel was set, an aliquot (5 µL) of the DNA was mixed with loading buffer (3 µL) plus water (7 µL) and loaded into the sample holes on the gel. Gels were run at 75 V for 3 h.
A Cary-Eclipse and a UV-Visible 5000 spectrophotometer (Varian, UK) were used to record the fluorescence-absorption spectra and melting temperature Tmin phosphate buffer (0.01 M phosphate, 0.1 M NaCl, at pH 7.4). Absorption and fluorescence spectra were recorded using 2 mL, thermostated, 4-sided quartz cuvettes. The mole ratio of oligonucleotides used was 1:1 (the concentration of each component was 2.5 µM in the cuvette). Most spectra were buffer-corrected for the particular temperature and particular wavelength region used. Melting curves were recorded by starting at a temperature well below the Tm and linearly increased the temperature well above the Tm (dissociation segment). The temperature was then decreased at the same rate until the starting temperature was reached
again (association segment). Tm experiments were directed and controlled by a computer with DNA melting software (Varian, UK). Melting curve experiments were performed spectrophotometrically and spectrofluorimetrically. Spectrophotometry was based on A260 profile, performed using the Cary 5000 UV-visible near IR spectrophotometer by measuring the change in absorbance at 260 nm with temperature. Spectrofluorimetrically determined melting curves were based on monitoring the change in relative fluorescence intensity with temperature at 650 nm (excitation wavelength) and 665 nm (emission wavelength) for the Cy5 at increasing temperature from 5 °C to 100 °C at a rate of 0.25 °C per minute. The sample was cooled at a rate of 0.25 °C per minute and the emission at the same wavelengths was monitored under similar conditions. The melting curve obtained during a cooling cycle was used to determine Tmvalues. The usual heating rate was 0.15 °C/minute. Values of Tm were defined as the maxima of the first-order derivatives of the melting curves and corresponded to within ± 1 °C to the values determined at half of the maximal hyperchromicity after baseline correction [17].
The Cary-Eclipse fluorescence spectrophotometer was used to record the emission/excitation spectra in phosphate buffer (0.01 M phosphate, 0.1 M NaCl, at pH 7.4). Emission spectra were recorded using 2 mL, thermostated, 4-sided quartz cuvettes. The excitation wavelength used was 650 nm and the emission was 665 nm for Cy5. Slit-widths were set from 2.5 nm to 5 nm, depending on the intensity of emission. The automatic shutter-on function was used to minimize photo-bleaching of the sample. All spectra were buffer-corrected for the particular temperature and particular wavelength region used.