Obsessive–Compulsive Disorder: Potential
Implications for Treatment Development
David R. Rosenberg and Gregory L. Hanna
In recent years, advances in brain research have resulted in a striking strategic shift in studies designed to develop new, effective treatments for neuropsychiatric disorders. This involves a multidisciplinary approach with recursive interactions among respective disciplines with the ultimate goal of contributing to treatment development. In this review we focus on treatment implications of brain imag-ing and molecular and pharmacogenetic studies in obses-sive– compulsive disorder. Translational components of this research are addressed, including the potential for integrating advances in brain imaging and molecular and pharmacogenetic assessments as they may potentially relate to neurodiagnostic assessment and treatment devel-opment. Studies of putative susceptibility alleles in obses-sive– compulsive disorder involving the serotonergic, glu-tamatergic, and dopaminergic systems may provide a focus for these divergent approaches. Taken together, neuroimaging and genetic methods may ultimately lead to a mechanistic understanding of the pathogenesis and maintenance of neuropsychiatric disorders such as obses-sive– compulsive disorder that may, in turn, result in the development of new neurodiagnostic and treatment ap-proaches. Biol Psychiatry 2000;48:1210 –1222 © 2000 Society of Biological Psychiatry
Key Words:Neuroimaging, genetics, pharmacogenetics, molecular genetics, obsessive compulsive disorders, brain
Introduction
U
nderstanding of the neurobiological and genetic un-derpinnings of major neuropsychiatric disorders has lagged behind the establishment of diagnostic criteria and treatment development for these conditions. Hyman(2000) argues persuasively that during the next decade psychiatry must adopt a multidisciplinary approach and integrate advances in neuroscience and genetics “to ask meaningful questions about the relationship among mind, brain, and behavior, and to finally overcome the pervasive Cartesianism that continues to incubate stigma and igno-rance about mental illness.” Brain imaging techniques in conjunction with advances in genetics can contribute a great deal to the outstanding questions in neuropsychiatry relevant to enhanced diagnostic rigor and treatment development.
Here we 1) review neurodiagnostic and treatment im-plications of recent brain imaging and molecular and pharmacogenetic studies relevant to pediatric obsessive– compulsive disorder (OCD) and 2) focus on the transla-tional components of this research—specifically, the po-tential for integrating advances in brain imaging and molecular and pharmacogenetics as they may relate to enhanced neurodiagnostic assessment and treatment de-velopment. Although OCD is used as a model for inter-facing brain imaging and genetic techniques, application to other neuropsychiatric disorders is clearly indicated.
Pediatric OCD is a severe, chronically disabling illness characterized by intrusive and distressing ritualistic thoughts and behaviors over which patients often have little if any control. Its lifetime prevalence is 2–3% (Flament et al 1988) and at least 80% of all cases of OCD have their initial onset in childhood and adolescence (Pauls et al 1995). Despite the advent of the selective serotonin reuptake inhibitors (SSRIs) and cognitive behav-ioral therapy (CBT), as many as one third of OCD patients do not respond at all to currently available treatment and many “responders” are only partial responders with con-tinued functional impairment (Grados et al 1999). This underscores how critical it is to develop new and more effective treatment approaches for this condition. As described below, taken together, neuroimaging and ge-netic methods may be especially useful in ultimately developing new biological treatments for neuropsychiatric disorders such as OCD.
From the Departments of Psychiatry & Behavioral Neurosciences and Pediatrics, Wayne State University, Detroit (DRR) and the Department of Psychiatry, University of Michigan, Ann Arbor (GH), Michigan.
Address reprint requests to David R. Rosenberg, M.D., Wayne State University School of Medicine, University Health Center, 9B-21, 4201 St. Antoine Boulevard, Detroit MI 48201.
Received March 3, 2000; revised June 6, 2000; accepted September 26, 2000.
Brain Imaging
Brain imaging techniques in conjunction with advances in developmental neurobiology, neuropsychopharmacology, and genetics can contribute a great deal to the outstanding questions regarding childhood-onset neuropsychiatric dis-orders such as OCD relevant to enhanced diagnostic rigor and treatment development. Neuroimaging techniques per-mit correlations between brain anatomy, chemistry, and function with baseline and outcome clinical assessment (e.g., Children’s Yale–Brown Obsessive–Compulsive Scale score, SSRI or CBT treatment) (Nemeroff et al 1999). As described below, neuroimaging studies have rather consistently implicated dysfunction within ventral prefrontal cortical (VPFC)–striatal–thalamic neural cir-cuits in OCD (Saxena et al 1998).
Treatment studies using biological markers may poten-tially lead to a mechanistic understanding of the pathogen-esis and maintenance of the illness, which may in turn result in the development of new diagnostic and treatment approaches. This is especially critical because OCD is a heterogeneous condition. The identification of particular abnormalities of brain anatomy, chemistry, and function might, for example, represent different etiologic or genetic forms of illness that may respond preferentially to differ-ent treatmdiffer-ent intervdiffer-entions. Brody et al (1998) have identified distinct patterns of regional brain glucose me-tabolism in adult OCD patients that predict differential treatment response to SSRIs versus CBT. Neuroimaging studies might identify specific neurobiological subtypes of OCD and help delineate the developmental neurobiologi-cal underpinnings of specific phenotypes of illness (Rauch 2000). This is likely to be important in view of the heterogeneity of the illness. Using brain imaging tech-niques that facilitate the noninvasive, longitudinal, and in vivo assessment of brain anatomy, chemistry, and function provides the advantages of minimizing potentially con-founding factors of longer term illness duration and treatment interventions and allows for a developmental framework for conceptualizing specific neuropsychiatric disorders such as OCD.
Identification of Neuroanatomic Structures
Involved in Pediatric OCD
Striatal Anatomy in Pediatric OCD
Neurobiological abnormalities in the striatum have been hypothesized to be the primary locus of pathology in OCD (Rauch et al 1998). Increased OCD behaviors are present in patients with basal ganglia disorders (for a review, see Fitzgerald et al 1999). Functional neuroimaging studies in adult OCD patients have demonstrated increased meta-bolic activity and brain activation in the caudate nucleus
correlated with severity of illness and response to treat-ment (Baxter et al 1992).
Structural neuroimaging studies in adult OCD patients, however, have demonstrated conflicting findings. Using computed tomography (CT), Insel et al (1983) found no difference between adult OCD patients and control sub-jects in ventricular brain ratios (VBRs). Of the six mag-netic resonance imaging (MRI) studies in adult OCD patients, two demonstrated abnormal caudate volumes (Robinson et al 1995; Scarone et al 1992), whereas four found no significant differences between OCD patients and control subjects (Aylward et al 1991, 1996; Bartha et al 1998; Kellner et al 1991). Such discrepancies may be related to different neuroimaging methodologies used in different studies, the heterogeneity of OCD, and poten-tially confounding factors such as comorbidity, illness duration, and most patients receiving psychotropic medication.
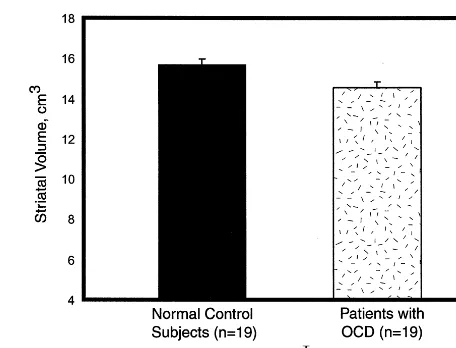
Interestingly, all published structural neuroimaging studies in pediatric OCD patients have detected abnormal-ities. Behar et al (1984) reported increased VBRs in adolescent OCD patients relative to control subjects. Although increased VBRs could be consistent with de-creased striatal volume, the authors did not report striatal measurements. Luxenberg et al (1988) used quantitative CT to demonstrate reduced caudate volumes in late ado-lescent and young adult male OCD patients. These reduc-tions were most prominent in the youngest male subjects in the sample. Rosenberg et al (1997c) identified reduced striatal volumes associated with OCD symptom severity but not illness duration in 19 treatment-naive OCD pa-tients, 7–17 years, versus 19 case-matched healthy com-parison subjects (Figure 1). No abnormalities were ob-served in case– control pairs in total brain volume.
Prefrontal Cortical Anatomy in Pediatric OCD
Disturbances in VPFC–striatal interactions are believed to be involved in causing OCD (for a review, see Fitzgerald et al 1999). Damage to VPFC circuits results in impair-ment in the ability to inhibit context-inappropriate emo-tional responses, leading to inappropriate behaviors (Stuss and Benson, 1983), whereas psychosurgical lesions of VPFC regions such as the anterior cingulate cortex can decrease OCD symptom severity in treatment-refractory patients (Chiocca and Martuza 1990). Eslinger and Damasio (1985) have reported that VPFC lesions cause disruption in “analysis and integration.” This results in impairment of the normal inhibitory control of VPFC circuits with consequent interruption of ongoing purposive behaviors in OCD patients (Flor-Henry et al 1979; Malloy 1987). The ventral prefrontal cortex is often referred to as the paralimbic cortex (Mesulam 1986) and through its connections with limbic regions in the cingulum and temporal lobes plays a critical role in integrating cognitive, motivational, and emotional mechanisms.
Using morphometric MRI, Robinson et al (1995) found no significant difference between total prefrontal cortical volume in adult OCD patients and control subjects. How-ever, measurement of localized prefrontal regions includ-ing the operculum (Jenike et al 1996) and orbitofrontal cortex (Szeszko et al 1998) has revealed abnormalities in adult OCD patients relative to control subjects that were correlated with severity of illness. More recently, Grachev et al (1998) found no difference between adult female OCD patients and matched control subjects in anterior cingulate, orbitofrontal, or opercular cortical volume. However, when they used a more sophisticated and accurate measurement technique, topographic parcellation
(Caviness et al 1996), six frontal parcellation units on the right and four parcellation units on the left were noted to be significantly increased in adult female OCD patients relative to matched female control subjects.
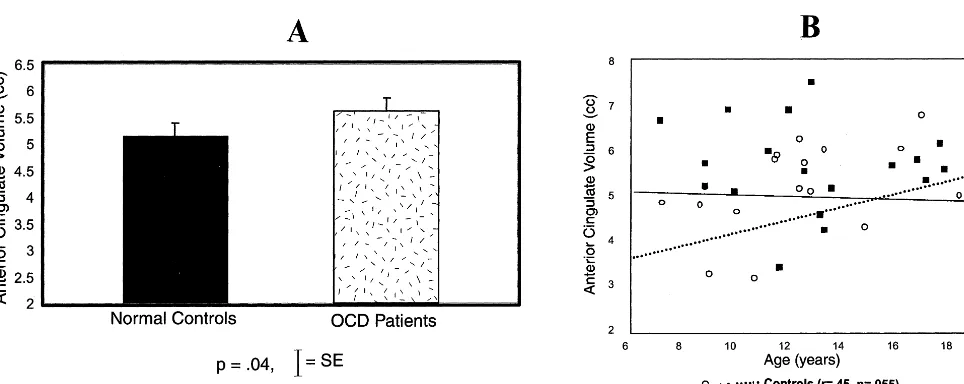
Consistent with Robinson and colleagues’ (1995) find-ing of no significant difference in total prefrontal cortical volume between adult OCD patients and control subjects, Rosenberg et al (1997c) failed to detect a significant difference in total prefrontal cortical volume between 19 treatment-naive pediatric OCD patients, 7–17 years, and case-matched control subjects. However, their subsequent studies of neurocognitive function (Rosenberg et al 1997a) and regional anatomy and signal intensity of the corpus callosum (MacMaster et al 1999; Rosenberg et al 1997b) pointed to abnormalities in the VPFC and suggested that measurement of total prefrontal cortical volume may have averaged out localized volumetric abnormalities in pedi-atric OCD patients. In a subsequent investigation, Rosen-berg and Keshavan (1998) reported that localized in-creased anterior cingulate volumes associated with OCD symptom severity but not duration of illness were ob-served in treatment-naive pediatric OCD patients versus healthy comparison subjects (Figure 2A). The normal age-related increase in anterior cingulate volume in healthy children was absent in OCD patients (Figure 2B). Increased anterior cingulate volumes were inversely cor-related with decreased striatal volumes in OCD patients.
Thalamic Anatomy in Pediatric OCD
Findings of increased VPFC volumes and reduced striatal volumes in OCD patients led Gilbert et al (2000) to study the thalamus, which serves as the final subcortical motor and sensory input to the frontal cortex, which stimulates
cortical output when released from the inhibitory tonic influence of the corpus striatum (Baxter et al 1996). Partial thalamotomy can reduce OCD symptoms in treatment-refractory OCD patients with severe manifestations of the illness (Chiocca and Martuza 1990). Functional neuroim-aging studies of adult OCD patients have also demon-strated increased metabolic rates and regional cerebral blood flow in the thalamus associated with OCD symptom severity and response to treatment (Baxter 1992).
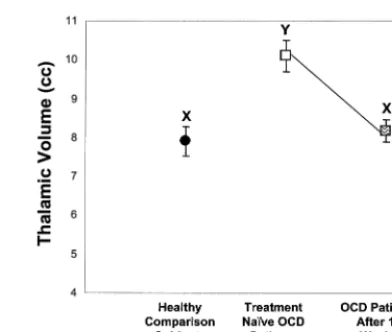
Although Jenike et al (1996) did not observe thalamic volumetric abnormalities between adult OCD patients and control subjects, many of the patients in this sample were on psychotropic medications. Using volumetric MRI, Gilbert et al (2000) reported increased thalamic volumes in 21 treatment-naive pediatric OCD patients relative to case-matched control subjects (Figure 3A). A differential maturation of thalamic volume was also observed in OCD
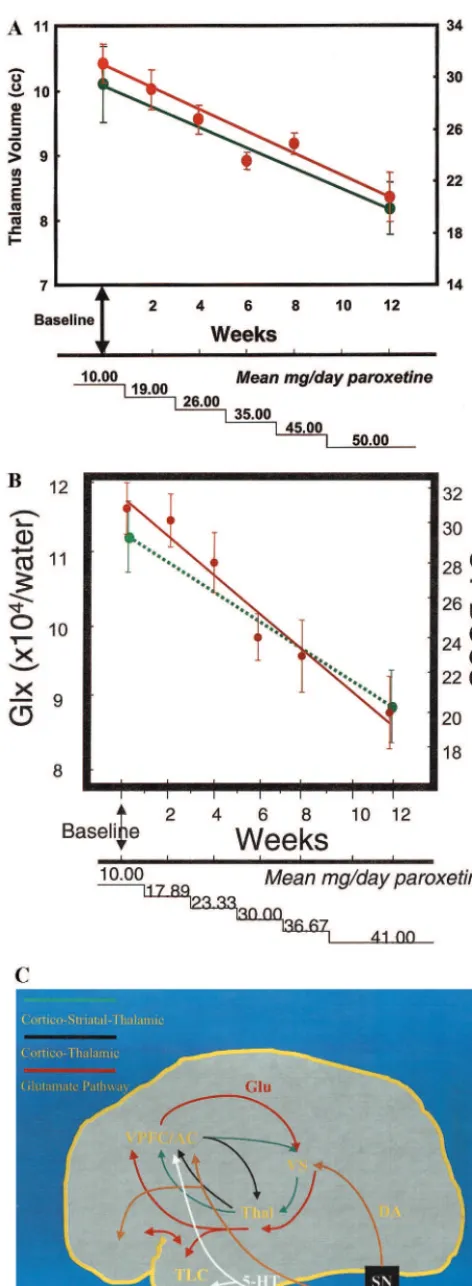
patients relative to control subjects (Figure 3B). Remark-ably, after 12 weeks of monodrug therapy with the SSRI paroxetine and no concomitant behavioral psychotherapy, there was a significant decrease in thalamic volume to levels comparable to and not significantly different from thalamic volumes in healthy children (Figure 4). Decrease in thalamic volume was positively correlated with de-crease in OCD symptom severity in pediatric OCD pa-tients (Figure 5A). In contrast, no significant differences were observed in 11 treatment-naive pediatric OCD pa-tients before and after CBT only (Rosenberg et al 2000a). No significant reductions in the VPFC, striatal volumes, or total brain volume were observed before and after parox-etine or CBT. Thus, localized reduction in thalamic volumes in pediatric OCD patients may be specific to treatment with the SSRI paroxetine as opposed to a general treatment effect and/or spontaneous remission.
Taken together, these results suggest that volumetric abnormalities in VPFC–striatal–thalamic circuitry may be associated with the clinical presentation of OCD and represent a central developmental neurobiological deficit in the illness. Abnormalities in thalamic anatomy, partic-ularly in younger children, may be reversible with effec-tive SSRI treatment but not with CBT. In contrast, abnormalities in VPFC and striatal volumes may not be reversible with effective SSRI or CBT treatment.
Increased anterior cingulate and thalamic volumes in pediatric OCD patients were consistent with prior obser-vations in childhood OCD patients of increased size and mylenization of the genu (MacMaster et al 1999; Rosen-berg et al 1997b), the region of the corpus callosum connecting the VPFC, and reports of increased metabolic rates in VPFC–striatal–thalamic circuitry in adult OCD patients (Baxter 1992). A growing body of evidence suggests that hyperactivity in the VPFC is directly linked
Figure 3. Thalamic volume by group(A)and thalamic volume versus age in pediatric obsessive– compulsive disorder (OCD) patients versus healthy comparison subjects(B).
to structures of the basal ganglia and thalamus in a parallel circuit that mediates the expression and manifestation of OCD symptoms (Rapoport and Wise 1988). Increased volume in the VPFC and thalamus in pediatric OCD patients may, however, represent an epiphenomenon re-lated to increased metabolic activity and cerebral blood flow in these brain regions. Differences in regional brain volumes between pediatric OCD patients and healthy control subjects does not necessarily indicate that these differences are pathogenic. They may, for example, rep-resent attempts to adapt or to compensate for the presence of obsessive thoughts or compulsive behaviors.
Since illness duration and age of onset of clinical OCD were not associated with anterior cingulate, thalamic, or striatal volumes, abnormalities in these regions are not consistent with a degenerative process in OCD but rather suggest that these abnormalities may be early neurobio-logical markers of the developmental pathogenesis of the illness. Ventral prefrontal cortical and striatal volumetric abnormalities may be biological markers associated with illness presentation, whereas thalamic volumetric abnor-malities may be reversible with effective SSRI treatment. It should be noted that the aforementioned studies involved relatively small sample sizes (21 OCD patients and 21 matched control subjects). Thus, these results must be considered preliminary, given the small sample size, and require replication. Refinements in measurement of regions, including the thalamus, whose reliabilities are difficult to measure are also critical for determining the underlying developmental neurobiology of OCD.
The aforementioned studies were also cross-sectional, so that degenerative processes cannot be excluded. Lon-gitudinal studies in affected OCD patients are currently ongoing in our laboratory and designed to monitor neuro-anatomic progression of illness over the course of illness as well as to determine whether neuroanatomic abnormal-ities persist after treatment discontinuation. Similar studies Figure 5. (A)Decrease in thalamic volume (green) associated with reduction in Obsessive–Compulsive score of the Children’s Yale–Brown Obsessive–Compulsive Scale (CY-BOCS; red).(B)
Left caudate glutamatergic concentrations versus obsessive– compulsive symptom severity as measured by the CY-BOCS versus paroxetine dosage. Green, glutamate/glutamine/g -ami-nobutyric acid (Glx); red, CY-BOCS.(C) Schematic diagram showing selected aspects of cortical–striatal–thalamic connec-tions in the neurodevelopment of obsessive– compulsive disor-der. Neurotransmitters: Glu, glutamate; DA, dopamine; 5-HT, serotonin. Brain regions: VPFC, ventral prefrontal cortex; AC, anterior cingulate; VS, ventral striatum; Thal, thalamus; TLC, temporal lobe cortex; RN, raphe nucleus; SN, substantia nigra; VT, ventral tegmentum.
are being conducted in offspring and siblings of OCD patients at increased risk for developing the illness.
The abnormalities in the normal developmental matu-ration of frontal–striatal–thalamic anatomy have been referred to as a “neural network dysplasia of OCD” (Rosenberg and Keshavan 1998). Such abnormalities may reflect disturbances in postnatal pruning, with increased pruning and resultant reduction in neural tissue in the striatum and decreased pruning resulting in increased neural brain elements in the VPFC and thalamus. Pruning of brain elements occurs during normal development (Keshavan et al 1994) but might be exaggerated in the striatum and reduced in the VPFC and thalamus.
The identification of potential critical windows of ab-normal maturation of VPFC–striatal–thalamic circuitry in pediatric OCD patients (Gilbert et al 2000; Rosenberg and Keshavan 1998) may, in part, explain why some studies of adult OCD patients have failed to detect comparable abnormalities. This is not meant to imply that OCD patients “grow out of the illness”; in fact, older OCD patients can also be significantly impaired. However, putative windows of aberrant maturation of particular brain regions may ultimately prove helpful in the identi-fication of differential treatment at different stages of development.
Brain Chemistry in Pediatric OCD
Rosenberg and Keshavan (1998) hypothesized that the frontal–striatal–thalamic abnormalities observed in pedi-atric OCD patients might involve neuronal dysfunction and abnormalities in glutamatergic tone impacting on serotonin circuits. Proton magnetic resonance spectros-copy (1H MRS) permits the direct, in vivo, and noninva-sive measurement ofN-acetyl-aspartate (NAA), a reliable marker of neuronal viability (Birken and Oldendorf 1989), and glutamatergic compounds (glutamate/glutamine/g -aminobutyric acid).
Neuronal Dysfunction in OCD
It has been hypothesized that the clinical symptomatology of OCD reflects impaired gating of sensorimotor informa-tion in VPFC–striatal–thalamic circuitry that results in the circuit’s firing continuously (Ebert et al 1997). The neu-ronal marker NAA, measured by 1H MRS, is localized primarily within neurons in the brain (Urenjak et al 1992). Decreased NAA levels are observed in diseases involving neuronal loss such as Alzheimer’s disease (MacKay et al 1996). Recent investigation suggests that reductions in NAA levels may represent an early marker of neuronal loss, dysfunction, or damage before such changes are detectable by structural MRI (Bartha et al 1998).
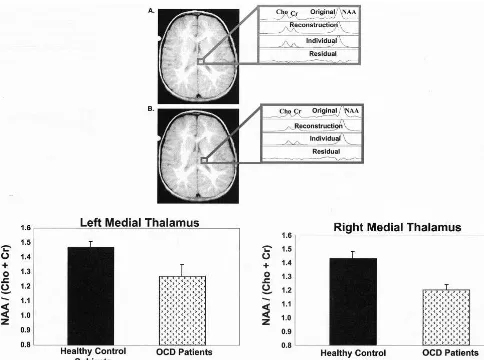
There-fore, NAA serves as a marker of both neuronal number and neuronal function/dysfunction (Tsai and Coyle 1995). Proton magnetic resonance spectroscopy studies have found decreased NAA levels in brain regions implicated in the pathophysiology of OCD, including the striatum (Bar-tha et al 1998; Ebert et al 1997) and VPFC (Ebert et al 1997) but not parietal cortex (Ebert et al 1997), a region not believed to be involved in the formation of OCD symptoms. Recently, Fitzgerald et al (2000) identified localized functional neurochemical marker abnormalities in the medial thalamus associated with OCD symptom severity but not illness duration. No abnormalities were observed in the lateral thalamus (Figure 6). This is an especially exciting finding, as the dorsomedial nucleus of the thalamus has been implicated in the pathophysiology of OCD (Modell et al 1989). It is important to point out that measurement of chemical spectra in the more medi-ally placed thalamic voxels may be confounded by possi-ble cerebrospinal fluid contamination. White matter con-tamination of more laterally placed thalamic voxels is also possible. It should be noted that controlling for thalamic volume confirmed significant differences between OCD patients and control subjects.
The previously reported pathologic correlations ob-served between VPFC–striatal–thalamic metabolic activ-ity in OCD patients (Baxter 1992) may be especially relevant in view of NAA abnormalities in OCD patients in these regions. Specifically, NAA abnormalities in VPFC– striatal–thalamic circuitry might result from persistent increased metabolic activity in these circuits. Glutamater-gic neurons project from the VPFC to the striatum and thalamus (Becquet et al 1990; Salt and Eaton 1996). Increased cortical–striatal–thalamic glutamate projections could lead to neurotoxicity and neuronal dysfunction.
Glutamate in Pediatric OCD
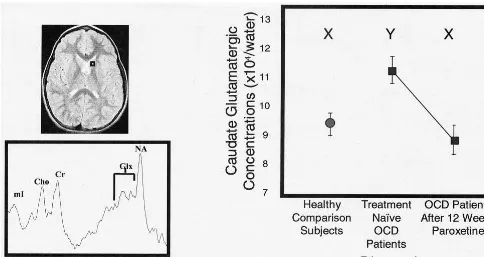
Using a short echo single-voxel 1H MRS approach, Rosenberg et al (2000b) observed significantly greater caudate glutamatergic concentrations in 11 pediatric OCD patients versus 11 case-matched healthy comparison sub-jects, with no case– control differences noted in occipital gray matter. Caudate glutamatergic concentrations de-creased significantly after 12 weeks of paroxetine mono-drug therapy to levels comparable to those observed in control subjects (Figure 7). Reduction in caudate glutama-tergic concentrations was positively correlated with reduc-tion in OCD symptom severity (Figure 5B). Increased pretreatment caudate glutamatergic concentrations in OCD patients predicted enhanced response to paroxetine intervention. Ongoing studies in our laboratory are fo-cused on examining glutamatergic concentrations in the VPFC and thalamus.
tergic compound projection with changes in VPFC–stria-tal–thalamic glutamate concentrations (Rosenberg et al 2000b).
In summary, this integrated series of brain imaging studies in pediatric OCD patients suggests a reversible glutamater-gically mediated thalamocortical–striatal dysfunction in OCD that may serve as a pathophysiologic and treatment response marker in pediatric OCD (Figure 5C). Critical neurodevelopmental changes in VPFC–striatal–thalamic cir-cuitry may be associated with the initial presentation of OCD. Treatment studies using biological outcome markers may lead to a mechanistic understanding of the pathogenesis and maintenance of the illness that may result in the development of new neurodiagnostic and treatment approaches. Neuroim-aging studies performed in conjunction with pharmacoge-netic and molecular gepharmacoge-netic studies (described below) may represent a unique and unprecedented opportunity for guid-ing a mechanistic understandguid-ing of neuropsychiatric disor-ders such as OCD and translating these advances into treatment development.
Potential Relevance of Molecular Genetic
and Pharmacogenetic Studies
The potential contributions of molecular genetic and pharmacogenetic strategies to neuroimaging studies and
treatment development in psychiatry have begun to re-ceive some discussion (Weinberger 2000; Weinshilboum 2000). It has been suggested that various neuroimaging techniques may be used to understand the biological expression of susceptibility genes for mental illness and to identify genetically determined responses to specific drugs. However, the recurrent difficulties in replicating findings of susceptibility loci for common psychiatric disorders preclude combining neuroimaging with most putative genetic markers in the near future. Instead, progress may be made by combining those strategies to investigate less common neuropsychiatric disorders—such as fragile X syndrome, Rett’s disorder, Prader–Willi syn-drome, and velocardiofacial syndrome— or to quantify the brain expression of genes with acknowledged behavioral effects (Lachman et al 1996; Lesch and Mo¨ssner 1998).
and dopaminergic systems may provide a focus for other-wise divergent genetic, neurobiological, and pharmaco-logic approaches to this complex brain disorder. The integration of genetic strategies into neuroimaging studies of early-onset OCD eventually may be rewarding because that form of the disorder appears to be highly familial (Hanna 2000; Nestadt et al 2000; Pauls et al 1995).
Studies have suggested that a functional polymorphism in the promoter region of the serotonin transporter (SLC6A4) is associated with anxiety-related traits. The initial study found that subjects with one or two copies of the short variant had higher neuroticism scores than did individuals homozygous for the long variant (Lesch et al 1996). In a subsequent study, linkage but not association was found between the polymorphism and anxiety-related traits, suggesting that the earlier finding may be related to another allele in the gene (Mazzanti et al 1998). Indeed, recent studies of the promoter region in the serotonin transporter gene revealed an internal fragment not reported in previous publications (Flattem and Blakely 2000; Mortensen et al 1999). This novel fragment contains consensus sequences for several transcription factors that require further study. The feasibility of examining the relationship between serotonin transporter genotype and in vivo protein expression has been demonstrated in a recent study of alcohol neurotoxicity (Heinz et al 2000).
As suggested by the studies of a possible relationship between the serotonin transporter gene and anxiety-related traits, the serotonin transporter is a plausible candidate gene for OCD and other psychiatric disorders responsive to potent serotonin reuptake inhibitors (SRIs). A study using the transmission disequilibrium test (TDT) provided preliminary evidence for association and linkage disequi-librium between the long allele in the serotonin transporter promoter region and OCD (McDougle et al 1998). There was also a trend for the long allele to be preferentially transmitted in the OCD patients who failed to respond to SRIs. However, case– control studies of the promoter polymorphism in OCD have yielded inconsistent results (Bengel et al 1999; Billett et al 1997). The larger of the two case– control studies found that patients with OCD were more likely to carry two copies of the long allele than matched control subjects (Bengel et al 1999). If the relationship between OCD and the serotonin transporter promoter polymorphism receives further confirmation with family-based association tests, it may be revealing to investigate the relationship between the polymorphism and brain expression of the gene in patients with OCD.
Two studies of depressed inpatients demonstrated that subjects with one or two copies of the long variant in the serotonin transporter promoter region had a better treat-ment response to fluvoxamine (Smeraldi et al 1998) and
paroxetine (Zanardi et al 2000). This is in contrast to the trend noted in the TDT study of the promoter polymor-phism in OCD (McDougle et al 1998). In another study of inpatients with bipolar depression, subjects homozygous for the long allele had a greater improvement in mood after total sleep deprivation (Benedetti et al 1999). It was suggested that having two copies of the long allele may allow more pronounced changes in serotonergic function-ing that are necessary for the antidepressant response to total sleep deprivation (Benedetti et al 1999; Hanna et al 1998). Further studies of the serotonin transporter pro-moter polymorphism may provide a better understanding of the wide variation in treatment response in OCD. The polymorphism may be particularly useful in predicting treatment response in the substantial number of OCD patients with comorbid depressive disorders (Hanna 1995; Welner et al 1976).
A preliminary genome scan of early-onset OCD with 65 individuals from seven families found maximum nonpara-metric LOD* scores above 1.5 on chromosomes 2, 9, and 17 (Hanna et al 1999). Although none of the results reached the level of statistical significance required for genetic linkage, the nonparametric LOD* score analyses suggested that three chromosomal regions may harbor susceptibility loci for early-onset OCD. The 2q region contains the serotonin 2B receptor gene (HTR2B; Le Coniat et al 1996). Activation of the 5-HT2B receptor in the rat causes anxiolysis and reduced grooming (Duxon et al 1997; Kennett et al 1997). No functional mutation was found, however, in the sequenced regions of the HTR2B in the probands from the families in the genome scan (Kim et al 2000).
9p monosomy has been previously associated with Tourette’s syndrome and OCD (Taylor et al 1991). The 9p region contains a neuronal glutamate transporter gene (SLC1A1; Smith et al 1993), which is provocative given the neuroimaging data previously described on the possi-ble role of glutamate in the pathogenesis of OCD. Again, no evidence was found for a functional mutation in the gene (Veenstra-VanderWeele et al, in press). Further linkage analyses with a larger number of families are warranted because other genes in the region may be considered positional candidates. Genome scans have the potential of identifying chromosomal regions likely to contain susceptibility genes for OCD without having a priorihypotheses about the pathogenesis of the disorder (Burmeister 1999). Given the previous problems with mapping genes for psychiatric and other common disor-ders, however, it may be possible to obtain consistent linkage findings with early-onset OCD only if it represents a more homogeneous form of the disorder (Hanna 2000; Nestadt et al 2000).
useful in understanding some aspects of the comorbidity or neurobiological heterogeneity of OCD. The distinction between tic-related and non–tic-related OCD has received support from phenomenological (Leckman et al 1995), natural history (Leonard et al 1992), neuroendocrine (Hanna et al 1991), pharmacologic (McDougle et al 1994), and family studies of OCD (Pauls et al 1995). A case– control study of the dopamine D4receptor gene in OCD patients found a higher rate of a seven-repeat allele in those patients with comorbid tics (Cruz et al 1997). Similarly, in a family-based association study of Tourette’s syndrome using the TDT, the seven-repeat allele of the D4receptor gene was preferentially transmit-ted (Grice et al 1996). Another case– control study found significant differences between OCD patients and matched control subjects in allele frequencies of a 48 – base pair repeat in the D4receptor gene (Billet et al 1998). Repli-cation with family-based association studies is required before any conclusions can be entertained about the role of the dopamine D4receptor gene in OCD. If an association is confirmed, genotypic information about the D4receptor may be productively integrated into neuroimaging or treatment studies of OCD.
Case– control and family-based association studies have implicated the genes for two enzymes involved in mono-amine metabolism, monomono-amine oxidase A (MAOA) and catecholO-methyltransferase (COMT), in the susceptibil-ity to OCD (Karayiorgou et al 1997, 1999). However, no linkage was found for a functional polymorphism in the COMT gene to Tourette’s syndrome and OCD (Barr et al 1999). As with the dopamine D4 receptor gene, further studies of the MAOA and COMT genes are needed to determine whether either gene is possibly associated with OCD or with another disorder that frequently coexists with OCD.
The genetic contribution to OCD is almost certainly complex, so current linkage and association techniques may be inadequate for mapping most of the important susceptibility loci for the disorder (Burmeister 1999; Hanna 2000). Gene chips and expression array technology hold promise for dealing with the genetic complexity of OCD and other common psychiatric disorders (Watson and Akil 1999). Assuming some progress in this area, each susceptibility gene may provide an option for treatment development. Advances in combinatorial chemistry and structure-based drug design have substantially increased the capacity to develop new drugs once targets are identified. Since the susceptibility genes for OCD may have to occur in a particular combination for symptoms to arise, it is possible that a drug targeting one of the susceptibility genes may be sufficient totreatthe disorder (Anderson and Cook 2000).
Conclusions
Rauch (2000) suggested that brain imaging profiles may provide biological phenotypes (“endophenotypes”) for genetic studies of OCD that are more etiologically homo-geneous than current clinical phenotypes and that the imaging profiles may be analyzed as either categoric or quantitative traits. Brain imaging profiles may be useful in clarifying the genetic heterogeneity of OCD or, more simply, in delineating biological differences between fa-milial and sporadic OCD. Yet it remains uncertain whether an advance in either neuroimaging or genetic studies of OCD will necessarily lead to an advance in the other area. Studies of common childhood neuropsychiatric disor-ders have rarely integrated genetic and neuroimaging strategies. A notable exception is the study by Castellanos and colleagues (1998) of attention-deficit/hyperactivity disorder. The cost of coupling a genome scan with neuroimaging is potentially prohibitive. However, if one or more candidate genes such as those described above are consistently associated with OCD, it may be become feasible to integrate genetic information into neuroimag-ing studies.
Neuroimaging studies may have their greatest value in psychiatry when they can be used to detail the brain expression of susceptibility genes for a complex neuro-psychiatric disorder such as OCD and to monitor the effects of biological and psychosocial treatments (Wein-berger 2000). Once susceptibility genes for OCD are identified, it should be possible to use pharmacogenomic techniques to develop more rational drugs for the disorder and to use neuroimaging techniques to further refine the effects of those treatments. Furthermore, expression array technology and pharmacologic imaging may have the potential to identify genetically determined responses to specific drugs.
This work was supported in part by the Joe Young Sr. Foundation and grants from the National Institute of Mental Health (Nos. MH01372 and MH59299), Rockville, Maryland, and the National OCD Foundation, Milford, Connecticut (DRR), and grants from the National Institutes of Health (Nos. MH56376 and MH01065) (GLH).
Aspects of this work were presented at the conference “Genetics and Brain Function: Implications for the Treatment of Anxiety,” March 22–23, 2000, Washington, DC. The conference was jointly sponsored by the Anxiety Disorders Association of America (ADAA), ADAA Scien-tific Advisory Board, and the National Institute of Mental Health.
References
Anderson GM, Cook EH (2000): Pharmacogenetics: Promise and potential in child and adolescent psychiatry.Child Adolesc Psychiatr Clin North Am9:23– 42.
obsessive-compulsive disorder assessed by quantitative neuroimaging.
Arch Gen Psychiatry53:577–584.
Aylward EH, Schwartz J, Machlin S, Pearlson G (1991): Bicau-date ratio as a measure of cauBicau-date volume on MR images.
Am J Neuroradiol12:1217–1222.
Barr CL, Wigg KG, Sandor P (1999): Catechol-O-methyltrans-ferase and Gilles de la Tourette syndrome.Mol Psychiatry
4:492– 495.
Bartha R, Stein MB, Williamson PC, Drost DJ, Neufield RW, Carr TJ, et al (1998): A short echo 1H spectroscopy and volumetric MRI study of the corpus striatum in patients with obsessive compulsive disorder and comparison subjects.Am J Psychiatry155:1584 –1591.
Baxter LR (1992): Neuroimaging studies of obsessive-compul-sive disorders.Psychiatr Clin North Am15:871– 884. Baxter LR, Saxena S, Brody AL, Ackermann RF, Colgan M,
Schwartz JM, et al (1996): Brain mediation of obsessive-compulsive disorder symptoms: Evidence from functional brain imaging studies in the human and nonhuman primate.
Semin Clin Neuropsychiatry1:32– 47.
Baxter LR, Schwartz JM, Bergman KS, Szuba MP, Guze BH, Mazziotta JC, et al (1992): Caudate glucose metabolic rate changes with both drug and behavior therapy for obsessive-compulsive disorder.Arch Gen Psychiatry49:681– 689. Becquet D, Faudon M, Hery F (1990): In vivo evidence for an
inhibitory glutamatergic control of serotonin release in the cat caudate nucleus: Involvement of GABA neurons.Brain Res
519:82– 88.
Behar D, Rapoport JL, Berg CJ, Denckla MB, Mann L, Cox C, et al (1984): Computerized tomography and neuropsycholog-ical test measures in adolescents with obsessive-compulsive disorder.Am J Psychiatry141:363–369.
Benedetti F, Serretti A, Colombo C, Campori E, Barbini B, de Bella D, et al (1999): Influence of a functional polymorphism within the promoter of the serotonin transporter gene on the effects of total sleep deprivation in bipolar depression.Am J Psychiatry156:1450 –1452.
Bengel D, Greenberg BD, Cora-Locatelli G, Altemus M, Heils A, Li Q, et al (1999): Association of the serotonin transporter promoter regulatory region polymorphism and obsessive-compulsive disorder.Mol Psychiatry4:463– 466.
Billet EA, Richter MA, Sam F, Swinson RP, Dai XY, King N, et al (1998): Investigation of dopamine system genes in obses-sive-compulsive disorder.Psychiatr Genet8:163–169. Billett EA, Richter MA, King N, Heils A, Lesch KP, Kenndy JL
(1997): Obsessive compulsive disorder, response to serotonin reuptake inhibitors and the serotonin transporter gene.Mol Psychiatry2:403– 405.
Birken DL, Oldendorf WH (1989): N-Acetyl-L-aspartic acid: A literature review of a compound prominent in 1H-NMR spec-troscopic studies of brain.Neurosci Biobehav Rev13:23–31. Brody AL, Saxena S, Schwartz JM, Stoessel PW, Maidment K,
Phelps ME, et al (1998): FDG-PET predictors of response to behavioral therapy versus pharmacotherapy in obsessive-compulsive disorder.Psychiatry Res84:1– 6.
Burmeister M (1999): Basic concepts in the study of diseases with complex genetics.Biol Psychiatry45:522–532. Calabresi P, Pisani A, Mercuri NB, Bernardi G (1996): The
corticostriatal projection: From synaptic plasticity to dysfunc-tion of the basal ganglia.Trends Neurosci19:279 –280.
Castellanos F, Lau E, Tayebi N, Lee P, Long R, Giedd J, et al (1998): Lack of an association between a dopamine-5 recep-tor polymorphism and attention-deficit/hyperactivity disor-der: Genetic and brain morphometric analyses.Mol Psychia-try3:431– 434.
Caviness VS Jr, Kennedy DN, Richelme C, Rademacher J, Filipek PA (1996): The human brain age 7–11 years: A volumetric analysis based on magnetic resonance images.
Cereb Cortex6:726 –736.
Chiocca EA, Martuza RL (1990): Neurosurgical therapy of obsessive compulsive disorder. In: Jenike MA, Baer L, Minichiello WE, editors. Obsessive-Compulsive Disorders: Theory and Management. Chicago: Year Book Medical Publishing, 283–294.
Cruz C, Camarena B, King N, Paez F, Sidenberg D, De la Fuente JR, et al (1997): Increased prevalence of the seven-repeat variant of the dopamine D4 receptor gene in patients with obsessive-compulsive disorder with tics.Neurosci Lett231:1– 4. Duxon MS, Kennett GA, Lightowler S, Blackburn TR, Fone
KCF (1997): Activation of 5-HT2B receptors in the medial amygdala causes anxiolysis in the social interaction test in the rat.Neuropharmacology37:265–272.
Ebert D, Speck O, Konig A, Berger M, Hennig J, Hohagen F (1997): 1-H-Magnetic resonance spectroscopy in obsessive-compulsive disorder: Evidence for neuronal loss in the cingulate gyrus and the right striatum.Psychiatry Res74:173–176. Edwards E, Hampton E, Ashby CR, Zhang J, Wang RY (1996):
5-HT3-like receptors in the rat medial prefrontal cortex: Further pharmacological characterization.Brain Res733:21–30. El Mansari M, Bouchard C, Blier P (1995): Alteration of
serotonin release in the guinea pig orbito-frontal cortex by selective serotonin reuptake inhibitors: Relevance to treat-ment of obsessive compulsive disorder. Neuropsychopharma-cology13:117–127.
Eslinger PJ, Damasio AR (1985): Severe disturbances of higher cognition after bilateral frontal lobe ablation: Patient EVR.
Neurology35:1731–1741.
Fitzgerald KD, MacMaster FP, Paulson LD, Rosenberg DR (1999): Neurobiology of childhood obsessive-compulsive disorder.Child Adolesc Psychiatr Clin North Am8:533–575. Fitzgerald KD, Moore GJ, Paulson LD, Stewart CM, Rosenberg DR (2000): Proton spectroscopic imaging of the thalamus in treatment-naive pediatric obsessive compulsive disorder.Biol Psychiatry47:174 –182.
Flament MF, Whitaker A, Rapoport JL (1988): Obsessive com-pulsive disorder in adolescence: An epidemiological study.
J Am Acad Child Adolesc Psychiatry27:764 –771.
Flattem NL, Blakely RD (2000): Modified structure of the human serotonin transporter promoter.Mol Psychiatry5:110 –115. Flor-Henry P, Yeudall LT, Koles ZJ, Howarth BG (1979):
Neuropsychological and power spectral EEG investigations of the obsessive-compulsive syndrome.Biol Psychiatry14: 119 –130.
Gilbert AR, Moore GJ, Keshavan MS, Paulson LD, Narula V, MacMaster FP, et al (2000): Decrease in thalamic volumes of pediatric obsessive compulsive disorder patients taking par-oxetine.Arch Gen Psychiatry57:449 – 456.
neocor-tex in obsessive-compulsive disorder.Arch Gen Psychiatry
55:181–182.
Grados M, Scahill L, Riddle MA (1999): Pharmacotherapy in children and adolescents with obsessive-compulsive disorder.
Child Adolesc Psychiatr Clin North Am8:617– 634. Grice DE, Leckman JF, Pauls DL, Kurlan R, Kidd KK, Pakstis AJ,
et al (1996): Linkage disequilibrium between an allele at the dopamine D4 receptor locus and Tourette syndrome, by the transmission-disequilibrium test.Am J Hum Genet59:644 – 652. Hanna GL (1995): Demographic and clinical features of obses-sive-compulsive disorder in children and adolescents.J Am Acad Child Adolesc Psychiatry34:19 –27.
Hanna GL (2000): Clinical and family-genetic studies of child-hood obsessive-compulsive disorder. In: Goodman WK, Ru-dorfer MV, Maser JD, editors.Obsessive-Compulsive Disor-der: Contemporary Issues in Treatment. Mahwah, NJ: Erlbaum, 87–103.
Hanna GL, Himle JA, Curtis GC, Koram DQ, Weele JVV, Leventhal BL, et al (1998): Serotonin transporter and seasonal variation in blood serotonin in families with obsessive-compul-sive disorder.Neuropsychopharmacology18:102–111. Hanna GL, McCracken JT, Cantwell DP (1991): Prolactin in
childhood obsessive-compulsive disorder: Clinical correlates and response to clomipramine. J Am Acad Child Adolesc Psychiatry30:173–178.
Hanna GL, Veenstra-VanderWeele J, Cox NJ, Boehnke M, Himle JA, Curtis GC, et al (1999): Genome scan of early-onset obsessive-compulsive disorder. In: Schwab-Stone ME, editor.The 46th Annual Meeting of the American Academy of Child & Adolescent Psychiatry.Washington, DC: American Academy of Child & Adolescent Psychiatry, 103.
Heinz A, Jones DW, Mazzanti C, Goldman D, Ragan P, Hommer D, et al (2000): A relationship between serotonin transporter genotype and in vivo protein expression and alcohol neuro-toxicity.Biol Psychiatry47:643– 649.
Hyman SE (2000): The millenium of mind, brain, and behavior.
Arch Gen Psychiatry57:88 – 89.
Insel TR, Donnelly EF, Lalakea ML, Alterman IS, Murphy DL (1983): Neurological and neuropsychological studies of pa-tients with obsessive-compulsive disorder. Biol Psychiatry
18:741–751.
Jenike MA, Breiter HC, Baer L, Kennedy DN, Savage CR, Olivares MJ, et al (1996): Cerebral structural abnormalities in obsessive-compulsive disorder: A quantitative morphometric magnetic resonance imaging study. Arch Gen Psychiatry
53:625– 632.
Karayiorgou M, Altemus M, Galke BL, Goldman D, Murphy DL, Ott J, et al (1997): Genotype determining low catechol-O -methyltransferase activity as a risk factor for obsessive-compul-sive disorder.Proc Natl Acad Sci U S A94:4572– 4575. Karayiorgou M, Sobin C, Blundell ML, Galke BL, Malinova L,
Goldberg P, et al (1999): Family-based association studies support a sexually diorphic effect of COMT and MAOA on genetic susceptibility to obsessive-compulsive disorder.Biol Psychiatry45:1178 –1189.
Kellner CH, Jolley RR, Holgate RC, Austin L, Lydiard RB, Laraia M, et al (1991): Brain MRI in obsessive-compulsive disorder.Psychiatry Res36:45– 49.
Kennett GA, Ainsworth K, Trail B, Blackburn TP (1997): BW
723C86, a 5-HT2B receptor agonist, causes hyperphagia and reduced grooming in rats.Neuropharmacology36:233–239. Keshavan MS, Anderson S, Pettegrew JW (1994): Is schizophrenia due to excessive synaptic pruning in prefrontal cortex? The Feinberg hypothesis revisited.J Psychiatry Res28:239 –265. Kim JS, Hassler R, Haug P, Paik KS (1977): Effect of frontal
cortex ablation on striatal glutamic acid level in rat.Brain Res
132:370 –374.
Kim S-J, Veenstra-VanderWeele J, Hanna GL, Gonen D, Lev-enthal BL, Cook EH (2000): Mutation screening of human 5-HT2B receptor gene in early-onset obsessive-compulsive disorder.Mol Cell Probes14:47–52.
Lachman HM, Papolos DF, Saito T, Yu YM, Szumlanski CL, Weinshilboum RM (1996): Human catechol-O-methyltrans-ferase pharmacogenetics: Description of a functional poly-morphism and its potential application to neuropsychiatric disorders.Pharmacogenetics6:243–250.
Leckman JF, Grice DE, Barr LC, deVries ALC, Martin C, Cohen DJ, et al (1995): Tic-related vs. non-tic-related obsessive compulsive disorder.Anxiety1:208 –215.
Le Coniat M, Choi DS, Maroteaux L, Launay JM, Berger R (1996): The 5-HT2B receptor gene maps to 2q36.3–2q37.
Genomics32:172–173.
Leonard HL, Lenane MC, Swedo SE, Rettew DC, Gershon ES, Rapoport JL (1992): Tics and Tourette’s disorder: A 2- to 7-year follow-up of 54 obsessive-compulsive children.Am J Psychiatry149:1244 –1251.
Lesch K-P, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al (1996): Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region.Science274:1527–1531.
Lesch K-P, Mo¨ssner R (1998): Genetically driven variation in serotonin uptake: Is there a link to affective spectrum, neurodevelopmental, and neurodegenerative disorders? Biol Psychiatry44:179 –192.
Luxenberg JS, Swedo SE, Flament MF, Friedland RP, Rapoport J, Rapoport SI (1988): Neuroanatomical abnormalities in obses-sive-compulsive disorder determined with quantitative x-ray computed tomography.Am J Psychiatry145:1089 –1093. MacKay S, Meyerhoff DJ, Constans JM (1996): Regional gray
and white matter metabolite differences in subjects with AD, with subcortical ischemic vascular dementia, and elderly controls with 1H magnetic resonance spectroscopic imaging.
Arch Neurol53:167–174.
MacMaster FP, Dick EL, Keshavan MS, Rosenberg DR (1999): Corpus callosal signal intensity in treatment naive pediatric obsessive compulsive disorder.Prog Neuropsychopharmacol Biol Psychiatry23:601– 612.
Malloy P (1987): Frontal lobe dysfunction in obsessive-compul-sive disorder. In: Perecman E, editor. The Frontal Lobes Revisited.New York: IRBN Press, 207–223.
Mazzanti CM, Lappalainen J, Long JC, Bengel D, Naukkarinen H, Eggert M, et al (1998): Role of the serotonin transporter promoter polymorphism in anxiety-related traits. Arch Gen Psychiatry55:936 –940.
McDougle CJ, Goodman WK, Leckman JF, Lee NC, Heninger GR, Price LH (1994): Haloperidol addition in fluvoxamine-refractory obsessive compulsive disorder: A double blind placebo-controlled study in patients with and without tics.
Arch Gen Psychiatry51:302–308.
Mesulam MM (1986): Frontal cortex and behavior.Ann Neurol
19:320 –325.
Modell JG, Mountz JM, Curtis GC, Greden JF (1989): Neuro-physiologic dysfunction in basal ganglia/limbic striatal and thalamocortical circuits as a pathogenetic mechanism of obsessive-compulsive disorder.J Neuropsychiatry1:27–36. Mortensen OV, Thomassen M, Larsen MB, Whittemore SR,
Wiborg O (1999): Functional analysis of a novel human serotonin transporter gene promoter in immortalized raphe cells.Brain Res Mol Brain Res68:141–148.
Nemeroff CB, Kilts CD, Berns GS (1999): Functional brain imag-ing: Twenty-first century phrenology or psychobiological ad-vances for the millenium?Am J Psychiatry156:671– 673. Nestadt G, Samuels J, Riddle M, Bienvenu J, Liang K-Y,
LaBuda M, et al (2000): A family study of obsessive-compulsive disorder.Arch Gen Psychiatry57:358 –363. Pauls DL, Alsobrook JP II, Phil M, Goodman W, Rasmussen S,
Leckman JF (1995): A family study of obsessive-compulsive disorder.Am J Psychiatry152:76 – 84.
Rapoport JL, Wise SP (1988): Obsessive-compulsive disorder: Evidence for basal ganglia dysfunction. Psychopharmacol Bull24:380 –384.
Rauch SL (2000): Neuroimaging research and the neurobiology of obsessive-compulsive disorder: Where do we go from here?Biol Psychiatry47:168 –70.
Rauch SL, Whalen PJ, Dougherty DD, Jenike MA (1998): Neurobiological models of obsessive compulsive disorders. In: Jenike MA, Baer L, Minichiello WE, editors.Obsessive Compulsive Disorders: Practical Management. Boston: Mosby, 222–253.
Reisine T, Soubrie P, Artaud F, Glowinski J (1982): Application of L-glutamic acid and substance P to the substantia nigra modulates in vivo [3H] serotonin release in the basal ganglia of the cat.Brain Res236:317–327.
Robinson D, Wu H, Munne RA, Ashtari M, Alvir JMJ, Lerner G, et al (1995): Reduced caudate nucleus volume in obsessive-compulsive disorder.Arch Gen Psychiatry52:393–398. Rosenberg DR, Averbach DH, O’Hearn KM, Seymour AB,
Birmaher B, Sweeney JA (1997a): Oculomotor response inhibition abnormalities in pediatric obsessive compulsive disorder.Arch Gen Psychiatry54:831– 838.
Rosenberg DR, Benazon NR, Gilbert AR, Lorch E, Moore GJ (2000a): Thalamic volume in pediatric obsessive compulsive disorder.Biol Psychiatry48:294 –300.
Rosenberg DR, Keshavan MS (1998): Toward a neurodevelop-mental model of obsessive compulsive disorder.Biol Psychi-atry43:623– 640.
Rosenberg DR, Keshavan MS, Dick EL, Bagwell WW, Mac-Master FP, Birmaher B (1997b): Corpus callosal morphology in treatment niave pediatric obsessive compulsive disorder.
Prog Neuropsychopharmacol Biol Psychiatry21:1269 –1283. Rosenberg DR, Keshavan MS, O’Hearn KM, Dick EL, Bagwell WW, Seymor AB, et al (1997c): Fronto-striatal measurement of treatment-naive pediatric obessive compulsive disorder.
Arch Gen Psychiatry54:824 – 830.
Rosenberg DR, MacMaster FP, Keshavan M, Fitzgerald KD, Stewart CM, Moore GJ (2000b): Decrease in caudate gluta-matergic concentrations in pediatric obsessive compulsive disorder patients taking paroxetine.J Am Acad Child Adolesc Psychiatry39:1096 –1103.
Salt TE, Eaton SA (1996): Functions of ionotropic and metabo-tropic glutamate receptors in sensory transmission in the mammalian thalamus.Prog Neurobiol48:55–72.
Saxena S, Brody AL, Schwartz JM, Baxter LR Jr (1998): Neuroimaging and frontal-subcortical circuitry in obsessive compulsive disorder.Br J Psychiatry173(suppl 35):26 –38. Scarone S, Colombo C, Livian S, Abbruzzese M, Ronchi P,
Locatelli M, et al (1992): Increased right caudate nucleus size in obsessive compulsive disorder: Detection with magnetic resonance imaging.Psychiatry Res45:115–121.
Sibson NR, Dhankhar A, Mason GF, Behar KL, Rothman DL, Shulman RG (1997): In vivo 13C NMR measurements of cerebral glutamine synthesis as evidence for glutamate-glu-tamine cycling.Proc Natl Acad Sci U S A94:2699 –2704. Smeraldi E, Zanardi R, Benedetti F, di Bella D, Perez J, Catalano
M (1998): Polymorphism within the promoter of the seroto-nin transporter gene and antidepressant efficacy of fluvoxam-ine.Mol Psychiatry3:508 –511.
Smith CP, Weremowicz S, Kanai Y, Stelzner M, Morton CC, Hediger MA (1993): Assignment of the gene coding for the human high-affinity glutamate transporter EAAC1 to 9p24: Potential role in dicarboxylic aminoaciduria and neurodegen-erative disorders.Genomics20:335–336.
Stuss DT, Benson DF (1983): Frontal lobe lesions and behavior. In: Kertesz A, editor.Localization in Neuropsychology.New York: Academic Press, 429 – 449.
Szeszko PR, Robinson D, Wu H, Ashtari M, Ma J, Alvir J, et al (1998): Decreased orbital frontal cortex volume in obsessive-compulsive disorder.Biol Psychiatry43:S20.
Taylor LD, Krizman DB, Jankovic J, Hayani A, Steuber PC, Greenberg F, et al (1991): 9p monosomy in a patient with Gilles de la Tourette’s syndrome.Neurology41:1513–1515. Tsai G, Coyle JT (1995): N-Acetylaspartate in neuropsychiatric
disorders.Prog Neurobiol46:531–540.
Urenjak J, Williams SR, Gadian DG, Noble M (1992): Specific expression of N-acetylaspartate in neurons, oligodendrocyte type 2 astrocyte progenitors, and immature oligodendrocytes in vitro.J Neurochem59:55– 61.
Veenstra-VanderWeele J, Kim S-J, Gonen D, Hanna GL, Lev-enthal BL, Cook EH (in press): Genomic organization of the
SLC1A1/EAAC1gene and mutation screening in early-onset obsessive-compulsive disorder.Mol Psychiatry.
Watson SW, Akil H (1999): Gene chips and arrays revealed: A primer on their power and their uses.Biol Psychiatry45:533–543. Weinberger DR (2000): Neuroimaging in the postgenome era.
Biol Psychiatry47:1S.
Weinshilboum R (2000): Pharmacogenomics. Biol Psychiatry
47:1S.
Welner A, Reich T, Robins L (1976): Obsessive compulsive neurosis: Record follow-up and family studies. I. Inpatient record study.Comp Psychiatry17:527–539.