Supplementary Material for:
MMFF VII. Characterization of MMFF94, MMFF94s, and Other
Widely Available Force Fields for Conformational Energies and for
Intermolecular-Interaction Energies and Geometries.
THOMAS A. HALGREN
Molecular Systems, Merck Research Laboratories, P.O. Box 2000, Rahway, NJ 07065
Appendix A: Experimental Conformational Energies for Set 1
Table A.I lists some, but by no means all, of the experimental conformational energies that
are available for the comparisons in Set 1. It also lists experimental uncertainties (a few of
which we estimated from data in the original report), indicates the type of quantity measured,
and specifies the method used. When two or more experimental values are listed, the first is
used in the comparisons of conformational energies made in this paper. As will be noted, in a
few instances we have chosen a different experimental value than the one we employed in our
earlier work.1 We also wish to point out that our earlier paper cited previous theoretical papers
as the basis for the listed values, whereas we now give citations to the original literature. For
conformational Set 2, we adopted the experimental values cited by Gundertofte2 without further
examination.
The table shows that the measured conformational energies form a disparate set. In
particular, many different experimental techniques have been used. More significantly, the
derived quantities sometimes refers to the gas phase and sometimes to the liquid phase or to
many incorrect experimental results. This fact is established simply by noting the many
instances in which alternative experimental values for the same conformational difference differ
by many multiples of the stated uncertainty in either. In such cases, both experimental results
cannot be correct (except possibly where strong medium effects are involved).
Given that some of the listed experimental values are wrong, it is fair to ask how a choice
can be made. One approach is to consider the reliability of the experimental technique. Our
own, somewhat untutored, prejudice is for determinations based on spectroscopic intensities
measured as a function of temperature, either directly (indicated by "/T" in the table) or on
samples cryogenically deposited from different initial temperatures (indicated by "/cdT"). In
such cases, a plot of the logarithm of relative intensity vs. 1/T gives ∆Η directly. Even this
technique, however, can give poor results, as the differences of more than 1 kcal/mol between
alternative experimental values determined in this way for 1,difluoroethane and for
2-butanone attest. We also favor determinations in which vibrational band progressions are used
to construct a complete torsional potential along a one-dimensional torsional coordinate. In
these cases, differences between local minima then give ∆E for that coordinate (though we call
this quantity ∆Η0 in recognition of the fact that the subsumed coordinates include zero-point and
thermal vibrational energy contributions), in close analogy to energy differences taken between
local minima on a force field or quantum mechanical energy surface. Conversely,
determinations based on conformationally averaged nmr chemical shifts or coupling constants
for which the isolated-conformer end-point values could not be measured directly seem to us
particularly prone to error.
The date of the measurement is also a factor, as we presume that more recent values are more
likely to be accurate, in part because of improved experimental technique but also because any
substantial variation from previously published values is likely to have been closely examined
by the authors. In some cases, however, our choice simply came down to which measurement
paper. Given their general success in predicting the observed conformational energies, we
presume that these very high level quantum calculations are unlikely to be seriously in error in
any individual case. To be sure, this choice minimizes the rms "error" reported for GVB-LMP2
(cf. Table IV), and its use might seem to compromise our claim that this method is the most
accurate. In point of fact, however, even selection of the conformational energies from Table
A.I in worst agreement with the GVB-LMP2 results still finds that it best reproduces the
experimental data. Moreover, we can think of no better approach for dealing with cases in
which discordant experimental values appear to have equal claims to validity. We had used
much the same approach previously,1 but with "MP4SDQ(T)/TZP" calculations serving as the
best reference values then available to us.
The reasons underlying most choices reflected in the table should be apparent from the
foregoing. Thus, we shall comment here only on a few cases. One of these is 2-butanone,
where all the higher-level calculations in Table III, as well as several others we had previously
reported,1 clearly favor the lower, liquid-phase result. A second case is 1,3-butadiene, where
we had cited a gauche - trans conformational energy difference of 2.5 kcal/mol, this being the
average of values referenced by Wiberg et al.4 This value, as it happens, also corresponds to an
often cited2,3b,5 but suspect early determination in which Carreira constructed a one-dimensional
torsional potential but used an erroneous torsional coordinate that incorrectly identified the
second, higher-energy conformer as being cis rather than gauche.22 The higher, more recent
values20,21 shown in Table A.I seem to us preferable. They are also consistent with the pattern
in Table III in which the theoretical method finds a larger gauche - trans difference for
1,3-butadiene than for 2-methyl-1,3-1,3-butadiene, evidently because the more compact trans conformer
in the methyl compound is somewhat destabilized by nonbonded repulsions involving the added
2-methyl group; Table I shows that MMFF94 and most of the other force-field methods show
In the case of 1-butene, we had previously cited a conformational energy difference of 0.53
kcal/mol.1 We now realize, however, that this value is derived from the free-energy difference
of 0.94 kcal/mol measured by Van Hemelrijk et al.65 by simply applying an entropic correction
of R ln2 = 1.18 e.u. to allow for the two-fold statistical factor favoring skew. However, the
looser skew conformation is also expected to have a higher vibrational entropy. With this mind,
we have applied the full entropic correction of 2.3 e.u. favoring skew found by Durig and
Compton64 to arrived at the value of 0.24 ± 0.42 kcal/mol listed in Table A.I. This correction to
the measured ∆G brings the derived value for ∆Η into line with Durig and Compton's value of
0.22 ± 0.02 kcal/mol and with the GVB-LMP2 result for ∆E.
The conformational energy difference for methyl vinyl ether is of particular interest. We
ourselves had previously referenced the Sullivan, Dickson, and Durig value of 1.70 kcal/mol
favoring cis over skew.49 Friesner and co-workers,3b however, cited the earlier Durig and
Compton difference of 1.15 kcal/mol,51 and worked intently to improve their theoretical
methodology to reduce the large discrepancy with their higher LMP2/cc-pVTZ(-f) theoretical
estimate of 2.62 kcal/mol.6 They subsequently published a GVB-LMP2 result of 1.45 kcal/mol
favoring skew.3b However, calculations using the current version of Jaguar now find the
conformational energy difference to be 2.14 kcal/mol (Table III, manuscript) when a rigorously
consistent protocol for lone-pair delocalization is employed in the localized MP2 calculation in
connection with improved grid and dealiasing functions. Furthermore, we find that this value
falls to 1.61 kcal/mol when zero-point energy differences3a are taken into account. This last
estimate is fully consistent with the Sullivan, Dickson, and Durig result and with the enthalpy
difference of 1.59 kcal/mol obtained Beech et al.,50 the experimental result that caused Durig to
reinvestigate this system and led him to raise his lower early estimate.
For the "simple" but conformationally critical gauche - anti conformational difference in n
-butane, we list what now appears to be a definitive value of 0.67 ± 0.10 kcal/mol.55 Also shown
0.75 kcal/mol value we had previously cited1 on the basis of Allinger, Yuh, and Li's
recommendation.7 Much as in the case of methyl vinyl ether, both the new value and one of the
earlier values it has superseded are by Durig and co-workers. Unfortunately, Durig et al. do not
discuss the reasons for rejecting their higher earlier value. However, they do cite a number of
other experimental determinations, made using a variety of methods, that agree with their lower
value of 0.67 kcal/mol.55 Other recent high-level quantum calculations8 also support this value,
as does the GVB-LMP2/cc-pVTZ(-f) value of 0.72 kcal/mol cited in Table III of the
manuscript.
As an indication of the limitations of experimental data, we point out that the cis - trans
difference for methyl acetate is particularly tenuous: the listed ∆G of 8.5 ± 1 kcal/mol represents
the relative intensity of a single weak band in the infra-red carbonyl region that was observed in
a sample cryogenically deposited from a temperature of 879 K and was thought to probably
arise from the cis conformer.14 Indeed, the original authors cite this conformational energy
difference as being a ∆Η, though this interpretation apparently assumes no entropy difference
between cis and trans conformers. We think it remarkable that the theoretical values in Table
III of the manuscript agree with the experimental value as well as they do, but caution that a
even a disagreement of 1 – 2 kcal/mol should not be viewed as indicative of a problem with the
theoretical calculation.
Finally, we discuss two entries for which we had previously based our comparisons on
free-energy differences measured in solution. First, we now list a single value of ∆G = 1.15
kcal/mol for the axial - equatorial difference in cyclohexyl amine rather than the previously
cited range of 1.1 - 1.8 kcal/mol.1 The reason for this change is that the larger values in the
range are for determinations in polar media that are inappropriate for comparison to calculated
isolated-molecule properties. Even this new reference value, however, is higher than the
theoretical calculations cited in Table III can accommodate. Second, we previously cited a free
enthalpy difference of 0.58 kcal/mol found by Eliel and Gilbert in dilute nonpolar medium for
the model 4-t-butylcyclohexanol system;46 these authors criticized the consensus value of 0.52
kcal/mol that Hirsch recommended for nonpolar solvents47 as being too low because it
incorporates low values for early determinations that they believed to be unreliable.
Most other minor differences between the present data and that cited previously1 either reflect
the substitution of representative specific determinations for previously referenced average
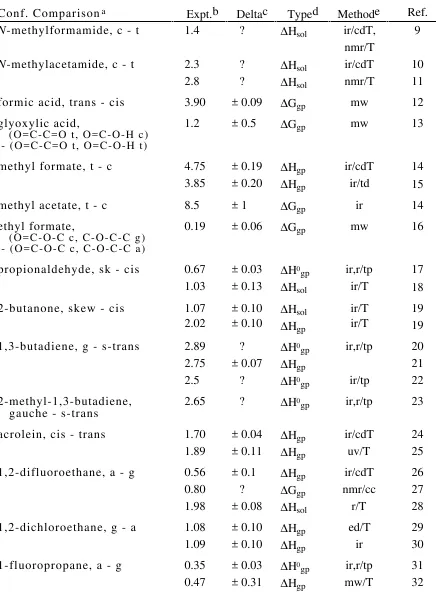
TABLE A.I.
___________________________________________________
References for Experimental Conformational Energies (Set 1), in kcal/mol.
Conf. Comparisona Expt.b Deltac Typed Methode Ref.
N-methylformamide, c - t 1.4 ? ∆Ηsol ir/cdT,
nmr/T
9
N-methylacetamide, c - t 2.3
2.8
methyl formate, t - c 4.75
3.85
propionaldehyde, sk - cis 0.67
1.03
2-butanone, skew - cis 1.07
2.02
1,3-butadiene, g - s-trans 2.89
2.75
acrolein, cis - trans 1.70
1.89
1,2-difluoroethane, a - g 0.56
0.80
1,2-dichloroethane, g - a 1.08
1.09
1-fluoropropane, a - g 0.35
1-chloropropane, g - a 0.09
ethanol, gauche - anti 0.12
0.3, 0.5
butane, gauche - anti 0.67
0.97
2,3-dimethylbutane,
1-butene, cis - skew 0.22
0.07
2-butene, cis - trans 1.2
1.0
a In the conformational notation, idealized dihedral angles are: cis or c, 0°; trans, t, anti, or a,
180°; gauche or g, 60°; and skew or sk, 120°.
b When more than one value for the conformational energy is cited, the first listed value is used
in the comparisons in the body of this paper
c Error in measurement as cited by the authors or as explained in a note accompanying the
literature citation.
d gp = gas phase; sol = solution or liquid-phase; superscript "0" in ∆Η0 denotes a difference
between the bottoms of potential wells constructed for a one-dimensional torsional coordinate, lT
means determined at low temperature.
e "r", "ir", "mw", "uv", nmr, and "ed" denote raman, infrared, microwave, ultraviolet, nuclear
magnetic resonance, and electron-diffraction spectroscopy; "/T" means as "a function of
temperature"; "/cdT" indicates sample collection via cryogenic deposition from thermalized
molecular beams as a function of temperature; "/tp" signifies construction of a one-dimensional
torsional potential; "mCal" stands for microcalorimetry; "nmr/cc" and "nmr/cs" denote nmr
determinations based on conformationally averaged chemical shifts and coupling constants,
respectively; "ir/td" stands for temperature-drift ir spectroscopy; and "comb." indicates
1. T. A. Halgren and R. B. Nachbar, J. Comput. Chem., 17, 587-615 (1996).
2. K. Gundertofte, T. Liljefors, P.-O. Norrby, and I. Petterssen, J. Comput Chem., 17, 429-449
(1996).
3. (a) R. A. Friesner, R. B. Murphy, M. D. Beachy, M. N. Ringnalda, W. T. Pollard, B. D.
Duneitz, and Y. Cao, J. Chem. Phys., submitted (1998); (b) R. B. Murphy, W. T. Pollard, and R.
A. Friesner, J. Chem. Phys., 106, 5073-5084 (1997).
4. K. B. Wiberg, P. R. Rablen, and M. Marquez, J. Am. Chem. Soc., 114, 8654-8668 (1992).
5. (a) N. L. Allinger, F. Li, L. Yan, and J. C. Tai, J. Comput. Chem., 11, 868-895 (1990); (b) C.
J. Casewit, K. S. Colwell, and A. K. Rappé, J. Am. Chem. Soc., 114, 10035-100046 (1992).
6. R. B. Murphy, M. D. Beachy, R. A. Friesner, and M. N. Ringnalda, J. Chem. Phys., 103,
1481-1490 (1995).
7. N. L. Allinger, Y. H. Yuh, and J.-H. Lii, J. Am Chem. Soc., 111, 8551-8566 (1989). See also
N. L. Allinger and L. Yan, J. Am. Chem. Soc., 115, 11918-11925 (1993), and references therein.
8. G. D. Smith and R. L. Jaffe. J. Phys. Chem., 100, 18718-18724 (1996).
9. Average of values in: S. Ataka, H. Takeuchi, and M. Tasumi, J. Mol. Struct., 113, 147-160
(1984), and F. A. L. Anet and M. Squillacote, J. Am. Chem. Soc., 97, 3243-3244 (1974).
10. S. Ataka, H. Takeuchi, and M. Tasumi, J. Mol. Struct., 113, 147-160 (1984).
11
.
T. Drakenberg and S. Forsén, Chem. Commun., 1404-1405 (1971). This result is from adifference in enthalpies of activation for isomerization in C2H4Cl2 as determined by nmr
line-shape analysis.
12. W. H. Hocking, Naturforsch., 31a, 1113-1121 (1976).
13. B. P. van Eijck and F. B. van Duijneveldt, J. Mol. Struct., 39, 157-163 (1977).
15. S. Ruschin and S. H. Bauer, J. Phys. Chem., 84, 3061-3064 (1980).
16. J. M. Riveros and E. B. Wilson Jr., J. Chem. Phys., 46, 4605-4612 (1967). This quantity is
reported to be the "energy difference" between the lowest vibrational levels, but its calculation
from measured microwave intensities at a single temperature suggests that it may be a
free-energy difference, presumably corrected for the two-fold statistical factor favoring the C-O-C-C
gauche conformer; no later report on this measurement appears to have been made.
17. J. R. Durig, D. A. C. Compton, and A. Q. McArver, J. Chem. Phys., 73, 719-724 (1980).
18. G. Sbrana and V. Schettino, J. Molec. Spectrosc., 33, 100-108 (1970). This value contributes
to the composite value of 0.95 kcal/mol cited in ref. 1. For other references, see: P. van Nuffel,
L. van den Enden, C. van Alsenoy, and H. J. Geise, J. Mol. Struct., 116, 99-118 (1984).
19. T. Shimanouchi, Y. Abe, and M. Makami, Spectrochim. Acta A, 24A, 1037-1053 (1968).
20. Y. N. Panchenko, A. V. Abramenkov and C. W. Bock, J. Mol. Struct., 140, 87-92 (1985).
21. J. R. Durig, W. E. Bucy, and A. R. H. Cole, Can. J. Phys., 53, 1832-1837 (1975).
22. L. Carreira, J. Chem. Phys.,62, 3851-3854 (1975).
23. Y. N. Panchenko, V. I. Pupyshev, A. V. Abramenkov, M. Traetteberg, and S. J. Cyvin, J.
Mol. Struct., 130, 355-359 (1985).
24. C. E. Blom and A. Bauder, Chem. Phys. Lett., 88, 55-58 (1982). Note that this value
supersedes the earlier result of 1.6 kcal/mol obtained by C. E. Blom, R. P. Müller and Hs. H.
Günthard, Chem. Phys. Lett., 73, 483-486 (1980).
25. E. J. Bair, W. Goetz, and D. A. Ramsay, Can. J. Phys., 49, 2710-2717 (1971). A note added
in proof to this paper reports a correction, agreed to by the original authors, to the value of 2.00 ±
0.11 kcal/mol published by: A. C. P. Alves, J. Christoffersen, and J. M. Hollas, Mol. Phys., 20,
26. P. Huber-Wälchi and Hs. H. Günthard, Chem. Phys. Lett., 30, 347-351. This result and the
cited uncertainty were obtained from the relative intensity vs. 1/T plots in Fig. 3. The authors
appear to incorrectly label their determinations as ∆G.
27. K. T. Hirano, S. Nonoyama, T. Miyajima, Y. Kurita, T. Kawamura, and H. Sato, J. Chem.
Soc., Chem. Commun., 1986, 606-607.
28. W. C. Harris, J. R. Holtzclaw, and V. F. Kalasinsky, J. Chem. Phys., 67, 3330-3338 (1977).
29. Average of values in Table 5 of: K. Kveseth, Acta Chem. Scand. A, 29, 307-311 (1975).
30. K. Tanabe, Spectrochim. Acta, 28A, 407-424 (1972). An entropy difference calculated from
symmetry numbers, moments of inertia, and vibrational frequencies was used to correct the
measured free-energy difference.
31. J. R. Durig, S. E. Godbey, and J. F. Sullivan, J. Chem. Phys., 80, 5983-5993 (1984).
32. E. Hirota, J. Chem. Phys., 37, 283-291 (1962).
33. W. A. Herrebout and B. J. van der Veken, J. Phys. Chem., 100, 9671-9677 (1996). I thank
Dr. Michael Beachy (Schrodinger) for supplying this reference.
34. K. Yamanouchi, M. Sugie, H. Takeo, C. Matsumura, and K. Kuchitsu, J. Phys. Chem., 88,
2315-2323 (1984). An entropy difference calculated from moments of inertia and vibrational
frequencies was used to correct the measured free-energy difference of -0.12 kcal/mol.
35. A. J. de Hoog, H. R. Buys, C. Altona, and E. Havinga, Tetrahedron, 25, 3365-3375 (1969).
Position of the equilibrium as a function of temperature was determined from conformationally
averaged nmr coupling constants.
36. R. M. Clay, G. M. Kelle, and F. G. Riddell, J. Am. Chem. Soc., 95, 4632 (1973).
37. J. R. Durig, G. A. Guirgis, and D. A. C. Compton, J. Phys. Chem., 83, 1313-1323 (1979).
39. Average of values for nonpolar solvents cited in H. Booth and M. L. Jozefowicz, J. Chem.
Soc., Perkins Trans. II, 895-901 (1976).
40. R. W. Baldock and A. R. Katritzky, J. Chem. Soc. B, 1470-1477 (1968); see also R. A. Y.
Jones, A. R. Katritzky, A. C. Richards, R. J. Wyatt, R. J. Bishop, and L. E. Sutton, J. Chem. Soc.
B, 127-131 (1970).
41. P. J. Crowley, M. J. T. Robinson, and M. G. Ward, Tetrahedron, 33, 915-925 (1977).
Kinetically-controlled protonation was used to trap conformers in quaternary salt form for nmr
analysis.
42. R. K. Kakar, and C. R. Quade, J. Chem. Phys., 72, 4300-4307 (1980).
43. J. R. Durig, W. E. Bucy, C. J. Wurrey, and L. A. Carriera, J. Phys. Chem., 79, 988-993
(1975). The authors note that the value chosen for ∆H depends on the choice for a key line
assignment.
44. H. L. Fang and R. L. Swofford, Chem. Phys. Lett., 105, 5-11 (1984). Signal detection was by
photoacoustic spectroscopy.
45. E. Hirota, as cited in W. A. Lathan, L. Radom, W. J. Hehre, and J. A. Pople, J. Am. Chem.
Soc., 95, 699-703 (1973). The reference to relative microwave intensities with no reference to
temperature dependence suggests that this measurement is ∆G, presumably corrected for the
two-fold statistical factor favoring the gauche conformer. For the preliminary study, see: S. Kondo
and E. Hirota, J. Mol. Spectrosc.,34, 97-107 1970). No follow-up study seems to have appeared.
46. E. L. Eliel and E. C. Gilbert, J. Am. Chem. Soc., 91, 5487-5495 (1969). The cited value was
measured for 4-t-butylcyclohexanol epimers equilibrated via Raney nickel catalysis.
47. J. A. Hirsch, in N. L. Allinger and E. L. Eliel, Eds., Topics in Stereochemistry, 1, 199-222
(1967). This often-cited value (cf. refs. 1, 2, 3b, and 5) is the free-energy difference Hirsch
recommends for nonpolar solvents, but has been criticized by Eliel and Gilbert46 as being too
48. T. Kitagawa and T. Miyazawa, Bull. Chem. Soc., Jpn., 41, 1976 (1968).
49. J. F. Sullivan, T. J. Dickson, and J. R. Durig, Spectrochim. Acta, 42A, 113-122 (1986).
50. T. Beech, R. Gunde, P. Felder, and Hs. Günthard, Spectrochim. Acta., 41A, 319-339 (1985).
51. J. R. Durig and D. A. C. Compton, J. Chem. Phys., 69, 2028-2935 (1978). Note that this
value is incorrect and has been superseded by the Sullivan, Dickson and Durig result.49
52. H. Weiser, W. G. Laidlaw, P. J. Kruger, and H. Fuhrer, Spectrochim. Acta, 24A, 1055-1089
(1968). The listed uncertainty of ± 0.05 reflects a purely subjective assessment on our part of the
data in Fig. 13 of this reference, and if anything is likely to be an overestimate.
53. F. R. Jensen, C. H. Bushweller, and B. H. Beck, J. Am. Chem. Soc.,91, 344-351 (1969).
54. E. L. Eliel and E. C. Gilbert, J. Am. Chem. Soc., 91, 5487-5495 (1969). This value was
measured for 4-t-butylmethoxycyclohexane using conformationally averaged nmr chemical
shifts.
55. W. A. Herrebout, B. J. van der Veken, A. Wang, and J. R. Durig, J. Phys. Chem., 99,
578-585 (1995). I thank Prof. Alex MacKerell (Univ. Maryland) for supplying this reference.
56. A. L. Verma, W. F. Murphy, and H. J. Bernstein, J. Chem. Phys., 60, 1540-1544 (1974).
57. J. R. Durig, A. Wang, W. Beshir, and T. S. Little, J. Raman. Spec., 22, 683-704 (1991).
58. R. K. Heenan and L. S. Bartell, J. Chem. Phys., 78, 1270-1274 (1983); W. F. Bradford, S.
Fitzwater, and L. S. Bartell, J. Molec. Struct., 38, 185-194 (1977). These papers are cited by
Allinger et al.7 as the basis for the value and the uncertainty listed in this table.
59. M. Squillacote, R. S. Sheridan, O. L. Chapman, and F. A. L. Anet, J. Am. Chem. Soc., 97,
3244-3246 (1975); this result uses data from: F. A. L. Anet and A. J. Bourn, J. Am. Chem. Soc.,
89, 760-768 (1967).
61. T. L. Boates, thesis, Iowa State University, 1966, as cited by: E. J. Jacob, H. B. Thompson,
and L. S. Bartell, J. Chem. Phys., 47, 3736-3753. This value is reported as a ∆G, but has been
corrected for the two-fold statistical factor in favor of the gauche conformer.
62. F. A. L. Anet and V. J. Basus, J. Am. Chem. Soc., 95, 4424-4426 (1973).
63. F. A. L. Anet and J. Krane, Isr. J. Chem., 20, 72-83 (1980). This result was measured by
nmr line-shape analysis at -145° and converted from ∆G = 0.5 kcal/mol by using a
force-field-calculated ∆S of 3.5 e.u. Use of the MP2/6-31G* estimate of 4.33 e.u. for ∆S (cf. ref. 1) would
raise ∆H by an additional 0.1 kcal/mol.
64. J. R. Durig and D. A. C. Compton, J. Phys. Chem., 84, 773-781 (1980). We estimated the
listed uncertainty of ± 0.02 kcal/mol from hand-drawn slopes for the five highest vs. four lowest
temperature points in Fig. 3 of this reference.
65. D. Van Hemelrijk, L. Van den Enden, H. J. Greise, H. L. Sellers, and L. Schäfer, J. Am.
Chem. Soc., 102, 2189-2195 (1980). The listed ∆H differs from the reported ∆H of 0.53
kcal/mol because it uses Durig and Compton's measured ∆S of 2.3 e.u. favoring skew (cf. ref. 64)
to make the full entropic correction (not just the symmetry-number correction of R ln2 = 1.18
e.u.) to the measured free-energy difference of 0.94 kcal/mol.
66. D. M. Golden, K. W. Egger, and S. W. Benson, J. Am. Chem. Soc., 86, 5416-5420 (1964).
67. Heat of combustion value cited in, but not correctly referenced by: N. L. Allinger, F. Li, and
L. Yan, J. Comput. Chem., 11, 848-867 (1990). The value listed here is from heats of formation
given in: Lange's Handbook of Chemistry, Fourteenth Edition, J. A. Dean, Ed., McGraw-Hill:
New York, NY, 1992. Measured heats of hydrogenation give similar values of 1.1 - 1.2
kcal/mol; see M. R. Ibrahim, Z. A. Fataftah, P. von R. Schleyer, and P. D. Stout, J. Comput.