1
Kelainan Genetik Sindrom Turner
Rudy Hermawan Cokro Handoyo 102010097-B2
Mahasiswa Fakultas Kedokteran Universitas Kristen Krida Wacana Jalan Terusan Arjuna Utara no. 6, Jakarta 11510
Email: [email protected]
Pendahuluan
Sindrom Truner adalah suatu sindroma pada perempuan yang terdiri dari postur pendek dan anomali kongenital mayor dan minor yang disebabkan kelainan kromosom seks. Biasanya pasien dengan sindrom turner datang dengan keluhan amenore dan infertilitas. Secara genetika telah kita ketahui bahwa jumlah kromosom pada genom manusia adalah 2n=46, yang terdiri dari 22 pasang autosom (22AA atau 44A) dan 2 kromosom seks (XX atau XY). Seorang perempuan mempunyai pasangan khromosom sex yang sama, yaitu khromosom X dan secara genetika ditulis 46,XX atau lebih singkat XX. Sebaliknya khromosom sex pada laki-laki merupakan pasangan tidak sejenis yaitu khromosom X dan Y dan ditulis 46,XY atau XY. Kadang terjadi gagal berpisah yaitu peristiwa tidak memisahnya kromosom selama pembalahan sel atau pada saat pembentukan gamet sehingga terbentuk mutan.1
Anamnesis
Penyelidikan pada kemungkinan penderita kelainan genetik dimulai dengan riwayat keluarga. Jika tidak bisa didapatkan anamnesis yang jelas dari pasien, maka dapat ditanyakan pada kerabat terdekat (orangtua, saudara, teman).Anamnesis dilakukan dengan bertanya:
1. Identitas pasien yaitu nama, tempat tanggal lahir, alamat, umur, suku, agama, pendidikan, dan pekerjaan, serta keadaan sosial ekonomi dan lingkungan tempat tinggalnya.
2. Menanyakan keluhan utama (amenore primer)
3. Riwayat penyakit sekarang (Diabetes Mellitus, Hipertensi, skoliosis) 4. Riwayat penyakit dahulu (pernah operasi karena kelainan jantung, ginjal) 5. Riwayat penyakit keluarga/ kecacatan:
Apakah kedua orang tua masih hidup ? ( jika masih hidup : usia berapa bagaimana kesehatannya , jika sudah meninggal : apa penyebabnya ? )
Berapa saudara kandung yang di miliki ? bagaimana keadaannya ?
Apakah ada anggota keluarga yang memiliki gejala yang sama atau mirip dengan pasien ?
2
Adakah anggota keluarga yang memiliki gejala klinis lain yang tidak sama dengan pasien, namun dapat menjadi ciri untuk diagnosa sementara untuk penyakit tersebut?
Adakah penyakit keturunan dalam keluarga yang sudah diketahui ?
Adakah anggota keluarga yang sakit atau meninggal karena penyakit langka ?
Adakah perkawinan dalam keluarga ?
Riwayat keluarga : misalnya pada kasus yang mengenai masalah tinggi badan, kita dapat menanyakan apakah dalam keluarga rata-rata memiliki tinggi badan yang sama? 6. Keadaan anak tersebut dulu saat dilahirkan dan perkembangannya (dari lahir sampai usia
sekarang)
7. Menanyakan umur ibu ketika mengandung pasien dan ANCnya.
Pemeriksaan Fisik
Pada pemeriksaan fisik kita dapat melakukan TTV karena pada sindrom turner biasanya ditemukan tanda-tanda hipertensi, kita bisa lakukan inspeksi dengan melihat perawakan anak ini cukup pendek umumnya tidak lebih dari 150 cm, bisa ditemukan web neck. Biasanya muka tampak lebih tua dari umurnya, epikantus positif, terdapat gangguan pertumbuhan dagu dimana akan mengganggu pertumbuhan gigi.
Dada berbentuk perisai (shield like chest) dengan putting susu yang letaknya berjauhan, terkadang ditemukan pektus eksavatum, kita juga bisa meihat apakah terdapat skoliosis, bisa dilakukan pemeriksaan jantung kebanyakan akan terdengar suara murmur karena coartasio aorta. Kita bisa temukan juga lymphedema pada kaki. Terdapat juga nevus pigmentosa pada kulit lebar.
Beberapa variasi yang bisa ditemukan:
Perawakan pendek (short stature)
Waktu lahir anak sudah pendek dan pada pertumbuhan selanjutnya biasanya berada di bawah persentil ketiga. Bila dewasa umumnya tidak lebih tinggi dari150 cm.
Webbed Neck
Merupakan lipatan kulit berbentuk segitiga (triangular skin fold ) yang terbentang dari telinga sampai ke akromion. Batas rambut biasanya rendah sekali, kadang-kadang sampai scapula.
Kelainan wajah
Muka biasanya agak aneh, tampak lebih tua dari umurnya. Ujung mata agak miring kebawah (anti mongoloid) dengan lipatan epikantus. Jarak interorbital agak kecil. Telinga besar dan rendah (low set ) dan kadang-kadang disertai cacat. Pertumbuhan dagu dan rahang bawah kurang sempurna sehingga tampak kecil. Hal ini juga akan mengakibatkan gangguan pertumbuhan gigi. Langit-langit melengkung tinggi.
3
Kelainan rangka
Hampir pada semua tipe ditemukan dada berbentuk perisai (shield-like chest ) dengan puting susu yang letaknya berjauhan. Kadang-kadang ditemukan pektus ekskavatus, gabungan beberapa tulang rusuk atau spina bifida, skoliosis, cubitus valgus, kelainan pergelangan tangan (pigeon chest Madrlung’s deformity) dan kelainan lutut. Sering tampak jari tangan dan kaki yang pendek dan kecil, sindaktili dan haluks valgus.
Kelainan kulit
Webbing di tempat lain seperti di jari dan aksila. Sering ditemukan keloid dan nevi
pigmentosa. Kadang-kadang ditemukan vitiligo. Cafeaulait spots dan neurofibromatosis Von Rlexklingshausen. Kuku sering kecil dan tipis (hipoplastik), letaknya dalam dengan pinggir kulit lebar.
Sistim kardiovaskular
Kira-kira 25% dari kumpulan kasus Wilkins disertai koarktasio aorta. Walaupun jarang, kadang ditemukan pula stenosis subaortik. Defek septum dan dekstro kardia, yang mungkin tidak ada hubungan langsung dengan sindrom ini. Hemangioma multipel dan leangiektasia pada usus pernah ditemukan. Limfedema sering di temukan pada bayi yang mempunyai sindrom Bonnevie-Uhrlich. Seringpula ditemukan hipertensi dengan sistolik berkisar antara 135-150 mmHg dan diastolic 90-110 mmHg, tetapi tidak didapatkan kecenderungan untuk naik. Tidak ditemukan kelainan ginjal, kecuali kadang-kadang didapatkan kelainan bawaan seperti horse shoe kidney.
Kelainan intelegensia
Sekitar ¼ - 1/3 dari penderita mempunyai kelemahan daya pikir. Terutama mereka yang mempunyai webbed neck . Di rumah sakit jiwa ditemukan 0,05% dari penderita wanita. Walaupun demikian didapatkan pula penderita yang cukup cerdas.
Kelainan pubertas
Karena kelainan gonad, anak tidak akan mengalami masa pubertas. Gejala seks sekunder yang karakteristik seperti pertumbuhan payudara, rambut aksila dan pubis serta perubahan labia minora tidak timbul atau hanya sedikit sekali. Semua hal ini sebagai akibat tidak adanya respons gonad untuk membuat estrogen.2
Pemeriksaan Penunjang
Analisis kromosom harus dipertimbangkan pada semua gadis pendek. Penderita dengan penanda kromosom pada beberapa atau semua sel harus diuji untuk rangkaian DNA pada atau dekat sentromer kromosom Y. Sebanyak 50% dari semua penderita 45,X mungkin membawa rangkaian Y, tetapi arti temuan ini belum diketahui. Hanya penderita dengan penanda kromosom
4
yang membawa rangkaian Y, yang memerlukan pengambilan gonad, karena mereka memiliki 30% risiko perkembangan gonadoblastoma.
Ultrasound jantung, ginjal, dan ovarium terindikasi setelah diagnosa ditegakkan. Kelainan skeleton yang paling lazim adalah pemendekkan tulang metakarpal dan metatarsal ke-4, disgenesis epifisis pada sendi lutut dan siku, deformitas Madelung, skoliosis, dan pada penderita yang lebih tua, mineralisasi tulang tidak cukup.
Kadar gonadotropin plasma, terutama hormon perangsang folikel (FSH) sangat meningkat di atas kadar kontrol sesuai umur selama masa bayi; pada usia sekitar 2-3 tahun, terjadi penurunan progresif pada kadarnya sampai kadar ini mencapai titik terendah (nadir) pada usia 6-8 tahun, dan pada usia 10-11 tahun, kadar ini meningkat sampai kadar dewasa kastrasi.
Antibodi antiperoksidasi thiroid harus dicek secara periodik, dan jika positif, kadar tiroksin dan hormon perangsang tiroid harus diukur. Penelitian yang luas telah gagal menentukan bahwa defisiensi hormon pertumbuhan memainkan peran primer dalam patigenesis gangguan pertumbuhan. Intoleransi karbohidrat ringan pada anak gadis muda cenderung membaik pada pubertas.3
Working Diagnosis
Sindrom Turner
Sindrome turner adalah satu-satunya monosomi yang memungkinkan kehidupan. Sindrome turner adalah aneuploidi tersering pada sejumlah abortus dan menyebabkan 20% dari keguguran pada trimester pertama. Prevalens adalah sekitar 1 dari 5000 kelahiran hidup. Terdapat 3 fenotip yang sering dijumpai pada 45 X. 98% hasil dari konsepsi mengalami kelainan berat sehingga cepat mengalami abortus. Fenotip kedua sering teridentifikasi oleh kelainan pemeriksaan sonografik yang mencakup higroma kistik, temuan-temuan ini sering disertai hidrops yang berkembang menjadi kematian janin. Fenotip ke tiga yang paling jarang dijumpai pada mereka yang lahir hidup.
Penemuan kromosom pada sindrom Turner merupakan kehilangan sebagian atau seluruh dari salah satu kromosom seks. Separuh individu yang terkena memiliki kromosom 45,X. Separuh lainnya memiliki berbagai kelainan kromosom seks. Fenotip pada sindrom ini adalah wanita. Sindrom Turner ditandai oleh badan pendek dan gonade yang tidak berkembang. Paling tidak separuh individu yang terkena mengalami edema perifer pada masa neonatus. Sepertiga individu yang terkena dikenali saat lahir karena adanya limfudem atau kulit ekstra atau jaringan leher, sepertiga dikenali saat masa kanak-kanak, biasanya karena badan pendek dan sepertiga tidak dikenali hingga mereka gagal memasuki pubertas karena displasia gonade. Sering ada malformasi kardiovaskuler (koarktasio aorta atau katup aorta bikuspid) dan ginjal. Tanda-tanda kelamin
5
sekunder tidak muncul pada 90% wanita yang terkena sehingga memerlukan pemberian hormon. Kebanyakan individu yang menderita sindrom Turner infertil. Biasanya intelegensinya normal, namun ada kesulitan dalam belajar. Diagnosis harus dilakukan lewat studi kromosom darah; studi pewarnaan mukosa pipi saja tidak cukup.4
Diferensial Diagnosis
Sindrom Insensitivitas Androgen
Sindrom insensitivitas androgen merupakan kelainan genetik resesif terkait X yang menghasilkan suatu spektrum fenotipe yang mengalami virilisasi tidak sempurna. Bentuk yang paling parah adalah AI (androgen insensifitas) komplet yang dahulu dikenal sebagai feminisasi testikular. Pada AI komplet, reseptor androgen intraselular tidak ada / tidak berfungsi. Induksi androgen terhadap perkembangan duktus wolfii tidak terjadi. Substansi penghambat mulleri dihasilkan oleh testis yang berfungsi normal sehingga duktus mulleri mengalami regresi. Testis turun sampai tingkat cincin inguinalis di bawah pengaruh MIS (mullerian inhibitting substance). Vagina yang pendek terbentuk dari sinus urogenital. Saat lahir, anak-anak dengan AI komplet biasanya ditetapkan memiliki jenis kelamin perempuan, karena tidak terdapat aktifitas androgen dan genitalia eksterna terlihat sebagai perempuan. AI komplet biasanya didiagnosis setelah pubertas ketika timbul gejala amenorea primer, dan adanya vagina pendek yang tidak berujung, tidak adanya serviks, uterus dan ovarium. Perkembangan payudara normal namun pertumbuhan rambut axila maupun pubis jarang.5
Sindrom insensitivitas androgen inkomplet (sindrom Reifenstein)
Jauh lebih jarang ditemukan dibandingkan dengan AL ( komplet dan berhubungan dengan spectrum fenotipe yang luas). Spektrum ini bervariasi dari kegagalan virilisasi genitalia interna dan eksterna yang hampir komplet hingga maskulinisasi fenotipe yang komplet. Diantara kedua keadaan yang ekstrim ini, terdapat pasien dengan klitoromegali ringan dan penyatuan labia yang tidak sempurna hingga pasien dengan ambigus genitalia yang signifikan. Baru-baru ini, beberapa pria yang digambarkan mengalami AL dengan indikasi infertilitas saja disebabkan oleh rendahnya atau tidak adanya produksi sperma. Beberapa pria subur yang mengalami masalah kejantanan memiliki bentuk yang ringan dari kelainan ini. AL inkomplet disebabkan oleh mutasi pada gen reseptor androgen. Gen yang mengkode reseptor androgen berada pada daerah q11-12 pada kromosom X. Defek dapat terjadi pada domain yang mengikat androgen pada reseptor, domain yang mengikat DNA reseptor, atau pada produksi protein reseptor. Kelainan yang diidentifikasi memiliki kisaran dari fungsi reseptor yang hilang total hingga perubahan kualitatif yang ringan pada transkripsi gen-gen target yang tergantung androgen. Terdapat korelasi yang lemah antara
6
kadar resptor androgen absolute dengan derajat maskulinisasi yang terlihat pada pasien dengan AL inkomplet.
Agenesis / Rokitansky
Agenesis vagina, yang juga dikenal sebagai Rokitansky sequence, diyakini terjadi akibat kegagalan duktus Mulleri bersatu dengan bagian posterior sinus urogenital dan seringkali disertai dengan tidak adanya uterus dan tuba uterina. Defek pada saluran kemih (45%) dan tulang belakang (10%) sering terjadi, disamping terdapat gangguan pendengaran. Pada pemeriksaan, terlihat cekungan tempat seharusnya terdapat pembukaan himen, dan genitalia eksterna lainnya tampak normal. USG biasanya menegaskan ketiadaan atau ketidaksempurnaan genitalia interna dengan ovarium normal. Hampir semua pasien mempunyai kariotipe 46,XX, tetapi pseudohermafrodit pada laki-laki harus disingkirkan dengan pencatatan kariotipik.
Terapi agenesis vagina adalah dengan membuat vagina ketika pasien menginginkan aktivitas seksual. Tindakan ini dapat dilakukan tanpa pembedahan dengan meminta pasien menggunakan serangkaian alat pelebar (dilator) dengan ukuran yang bertambah besar secara progesif untuk menghasilkan tekanan konstan pada cekungan, tempat seharusnya terdapat himen, selama 20-30 menit setiap hari selama beberapa bulan. Jika tindakan ini tidak berhasil, vagina dapat dibuat dengan pembedahan.
Agenesis vagina parsial, yang biasanya hanya pada sepertiga bagian bawah, diyakini terjadi
akibat kegagalan epitel sinus urogenital menembus vagina pada kehamilan 4-5 bulan. Vagina bagian atas, uterus, dan tuba normal. Pemeriksaan rektum dapat menunjukkan vagina bagian atas yang membesar (terutama pada pasca menarke), dan dapat dijumpai kelainan ginjal.
Terapi agenesis vagina parsial mengharuskan adanya drainase vagina bagian atas yang terobstruksi, biasanya dengan membuat vagina bagian bawah.5
Etiologi
Sindroma Turner disebabkan karena terjadi nondisjunction pada proses mitosis ataupun miosis serta usia ibu diatas 35 tahun. Karena pada usia tersebut terjadi tahapan arrest pada profase 1 yang panjang di oogenesis sehingga pada waktu segregasi (pemisahan diri) terjadi kegagalan.6
Epidemiologi
Resiko terhadap sindrom tidak meningkat sejalan dengan usia ibu. Sindrom turner (Disgenesis
gonad) 60.000 mempengaruhi perempuan di Amerika Serikat. Gangguan ini terlihat dalam 1 dari
setiap 2000-2500 bayi lahir, dengan sekitar 800 kasus baru didiagnosa setiap tahun. Pada 75-80% kasus, satu kromosom X berasal dari telur ibu, sperma ayah yang menyuburkan telur hilang dengan seks kromosom. Sindrom turner (Disgenesis gonad) bukan merupakan penyakit keturunan tetapi kadang diduga salah satu orang tua membawa kromosom yang telah mengalami
7
penyusunan ulang, yang bisa menyebabkan terjadinya sindrom ini. Kehilangan suatu kromosom seks(45,x) sering ditemukan pada jaringan-jaringan abortus dan hanya sekitar 3-5% fetus dengan sindrom turner yang dapat mencapai trimester ketiga kehamilan.6
Patofisiologi
Monosomi X seperti halnya sindroma Turner ini mungkin terjadi karena adanya nondisjunction diwaktu ibunya membentuk sel telur. Kemungkinan lain disebabkan karena hilangnya sebuah kromosom kelamin selama mitosis setelah zigot XX atau XY terbentuk. Kemungkinan yang terakhir ini didukung oleh tingginya frekuensi mosaic yang dihasilkan dari kejadian sesudah terbentuk zigot pada penderita Turner. Mosaik dengan kromosom kelamin X/XX memperlihatkan tanda-tanda sindroma Turner, tetapi biasanya orangnya lebih tinggi daripada X dan mempunyai lebih sedikit anomali daripada wanita non-mosaik 45,X. mereka lebih memperhatikan kewanitaannya, mempunyai siklus haid lebih kearah normal dan mungkin subur. Kini banyak dijumpai kasus fenotipe Turner somatic tanpa disertai kombinasi kromosom 45,X. kebanyakan dari pasien ini memiliki sebuah kromosom-X normal dan sebuah potongan dari kromosom-X yang kedua. Kedua buah lengan dari kromosom-X yang ke dua rupa-rupanya sangat diperlukan untuk differensiasi ovarium secara normal. Individu yang hanya memiliki lengan panjang dari kromosom-X ke dua, mempunyai tubuh pendek dan menunjukkan tanda-tanda lain dari sindroma Turner. Mereka yang hanya memiliki lengan pendek dari kromosom-X yang ke dua, mempunyai tubuh normal dan tidak menunjukkan banyak tanda-tanda sindroma Turner. Pendapat baru inilah memberi kesan bahwa fenotipe Turner itu diawasi oleh gen-gen yang terdapat dalam lengan pendek dari kromosom-X.
Hasil penelitian lain yang menarik perhatian pula dapat diterangkan sebagai berikut. Pasien yang kehilangan sebagian dari kromosom-X adalah seks kromatin positif dan berhubung dengan itu dapat mengakibatkan kekeliruan dalam diagnose bila pemeriksaan hanya dilakukan dengan tes seks kromatin saja. Kromosom-X yang mengalami defisiensi selalu membentuk seks kromatin. Pada beberapa individu dengan fenotipe Turner terdapat pula sebuah kromosom-Y. Pasien-pasien demikian ini biasanya mosaic untuk 45,X/46,XY dengan sebuah kromosom-Y normal. Orang dengan sebuah kromosom-X dan sebuah potongan kromosom-Y, tidak termasuk lengan pendek dari kromosom-Y, hanya memiliki goresan ovarium, tetapi mempunyai fenotipe normal. Hal ini memberi petunjuk bahwa gen-gen yang menentukan jantan terhadap lengan pendaek dari kromosom-Y.
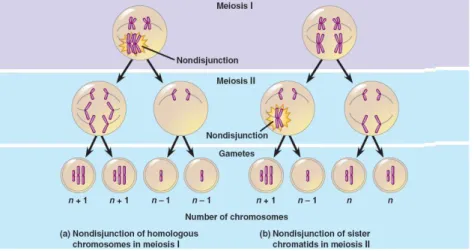
Mekanisme non-disjunction pada fase miosis 1 / miosis 2 di sel gamet (ovum/sperma), kelainan yang disebabkan oleh mekanisme ini akan berakibat trisomi, tetrasomi atau monosomi pada semua sel. Aneuploidi yang disebabkan oleh non-disjunction pada fase miosis umumnya
8
menyebabkan abortus, kematian janin, atau kecacatan berat sehingga bayi tidak bertahan hidup lama. Aneuploidi yang sering dijumpai adalah sindrom down kromosom 21 dan sindrom turner monosomi X bisa juga kelainan jumlah kromosom seks seperti XXY, XXX, XYY, karena aneuploidi ini bisa bertahan hidup sehingga dapat ditemui di klinik.
Penyebab non disjunction fase miosis lebih dihubungkan dengan usia lanjut ibu pada saat hamil. Jika pada fase mitosis (post zygotiv non-disjunction) tergantung atas fasenya yaitu pada sel pertama zigot atau setelah mitosis zigot makajenis kelainan kromosom bisa mosaik sel dengan kromosom trisomi dan monosomi bila terjadi pada sel pertama atau mosaik sel dengan kromosom normal (diploid) sel trisomi dan monosomi bila terjadi setelah mitosis setelah mitosis normal terjadi beberapa tahap.2
Gambar 1. Non-disjunction in miosis 1 & 2
sumber : http//:bio1151.nicerweb.com
Gejala Klinis
Dahulu, diagnosis biasanya mula-mula dicurigai pada masa anak atau pada pubertas ketika maturasi seksual gagal terjadi. Banyak penderita dengan sindrom Turner dapat dikenali pada saat lahir karena edema khas dorsum tangan dan kaki dan lipatan kulit longar pada tengkuk leher. Berat badan lahir sangat rendah dan panjang badan yang kurang lazim. Manifestasi klinis pada masa anak meliputi selaput pada leher, batas rambut posterior rendah, mandibula kecil, telinga menonjol, lipatan epikantus, lengkungan palatum tinggi, dada lebar yang memberikan penglihatan yang salah puting yang sangat lebar, kubitum valgum, dan kuku jari sangat cembung.
Perawakan pendek, temuan utama pada semua gadis dengan sindrom Turner, mungkin datang dengan manifestasi klinis lain minimal. Selama umur tiga tahun pertama, kecepatan pertumbuhan normal, meskipun pada persentil yang lebih rendah; setelahnya pertumbuhan mulai melambat dan menghasilkan perawakan yang sangat pendek. Maturasi seksual gagal terjadi pada usia yang
9
diharapkan. Rata-rata tinggi badan dewasa adalah 143 cm (132-155 cm). Nevi berpigmen semakin tua menjadi lebih nyata.
Defek tersembunyi yang menyertai lazim ada. Evaluasi jantung lengkap, termasuk ekokardiografi, menampakkan katup aorta bikuspid nonstenotik murni pada sekitar sepertiga penderita. Defek yang kurang sering tetapi lebih serius adalah stenosis aorta, koarktasio aorta, dan anomali muara vena pulmonalis. Sekitar sepertiga penderita menderita malformasi ginjal pada pemeriksaan ultrasuara. Defek yang lebih serius adalah ginjal pelvis, ginjal bentuk sepatu kuda, sistem kolektivus ganda, satu ginjal tidak ada sama sekali, dan obstruksi sambungan ureteropelvis.
Bila ovarium diperiksa dengan ultrasound, ovarium kecil tetapi tidak bergaris-garis ditemukan pada setengah dari penderita pada umur 4 tahun pertama; antara usia 4 dan 10 tahun, ovarium tampak bergaris pada 90% pendereita. Maturasi seksual biasanya gagal terjadi, tetapi 10-20% wanita secara spontan mengalami perkembangan payudara, dan bahkan kadang-kadang wanita dapat mengalami beberapa masa menstruasi. Lebih dari 50 kehamilan telah dilaporkan pada penderita dengan sindrom Turner yang mengalami menstruasi secara spontan.
Otitis media bilateral kambuhan terjadi pada sekitar 75% penderita. Defisit pendengaran sensorineural lazim ada, dan semakin tua frekuensinya semakin meningkat. Masalah meningkat dengan integrasi motor-sensoris kasar dan halus, tidak berhasil berjalan sebelum usia 15 bulan, dan disfungsi kemampuan berbicara dini sering mengundang tanda tanya mengenai keterlambatan perkembangan, tetapi inteligensia normal. Namun retardasi mental terjadi pada penderita dengan 45,X/46,X,r(X), karena cincin kromosom tidak mampu melakukan inaktivasi dan menyebabkan dua kromosom X berfungsi. Pada orang dewasa, defisit pada kemampuan persepsi ruang lebih lazim daripada mereka yang dari populasi umum.
Adanya gondok akan memberi kesan tiroiditis limfositik. Nyeri perut, tenesmus, atau diare berdarah dapat menunjukkan adanya penyakit radang usus; dan pendarahan seluran pencernaan kambuhan dapat menunjukkan telangiektasia saluran pencernaan.
Pada penderita dengan mosaikisme 45,X/46,XX, kelainannya diperlemah dan lebih sedikit; perawakan pendek adalah sama seringnya dengan perawakan pendek pada penderita 45,X dan dapat hanya merupakan manifestasi dari keadaan selain dari kegagalan ovarium.5
Penatalaksanaan
1. Segi psikologi
Terhadap anak harus diyakinin sedemikian rupa sehingga ia mempunyai perasaan seperti anak wanita lainnya yang seumur. Untuk itu perlu diberikan hormon kelamin dan terhadap orangtua perlu diberikan keyakinan bahwa terapi hormonal ini diperlukan.
10
Dimulai kalau sudah akil-balik. Sebaiknya lebih dahulu dilakukan pemeriksaan kadar gonodotropin penderita. Diberikan hormon estrogen terus menerus selama 6-9 bulan sehingga timbul pertumbuhan payudara, vagina dan uterus. Sesudah masa ini, estrogen dapat diberikan secara siklik, yaitu selama 21 hari dan 2-5 hari kemudian akan timbul menstruasi. Kalau respons terhadap estrogen kurang baik, dapat ditambahkan progesteron selama minggu ketiga dari siklus tersebut. Data menunjukan pengobatan dengan hormone pertumbuhan rekombinan saja atau bersama dengan steroid anabolik meningkatkan kecepatan tinggi badan. Banyak gadis dapat mencapai tinggi badan 150 cm atau lebih dengan memulai pengobatan dini.
Terapi penggantian dengan estrogen terindikasi, tetapi ada sedikit kesepakatan mengenai usia optimal memulai pengobatan. Kesiapan psikologis penderita untuk mendapatkan terapi harus diperhitungkan. Dahulu ada kecenderungan untuk menunda terapi penggantian dengan estrogen untuk mencapai tinggi badan maksimal. Pertumbuhan membaik yang dicapai oleh gadis yang diobati dengan hormon pertumbuhan memungkinkan memulai penggantian estrogen pada usia 12-13 tahun. Premarin , 0,3-0,625 mg yang diberikan setiap hari selama 3-6 bulan, biasanya efektif untuk menginduksi pubertas. Estrogen kemudian diputar (diminum pada hari 1-23), dan Proovera, suatu progestin, ditambahkan (diminum pada hari 10-23) dengan dosis 5-10 mg per hari. Pada sisa bulan kalender, selama waktu tersebut tidak diberikan pengobatan, pendarahan penarikan (withdrawal) biasanya terjadi. Sediaan estrogen lain dan regimen pengobatan sekarang juga digunakan.
Analisis kromosom prenatal pada usia ibu yang sudah lanjut telah menyingkap frekuensi 45,X/46,XX yaitu 10 kali lebih tinggi daripada ketika didiagnosis pascanatal. Kebanyakan penderita ini tidak memiliki manifestasi klinis sindrom Turner, dan kadar gonadotropin normal. Menyadari fenotip ringan ini penting dalam memberikan nasehat penderita.
Dukungan psikososial terhadap gadis-gadis ini merupakan komponen penting pada penanganan. Organisasi sindrom Turner, yang memiliki cabang lokal di Amerika Serikat, dan kelompok serupa di Kanada dan negara-negara lain memberikan sistem dukungan yang berharga pada penderita-penderita ini dan keluarganya disamping yang diberikan oleh dokternya.5
Prognosis
Prediksi hasil akhir penyakit atau perkembangan jangka panjang seorang anak merupakan pendekatan terbaik. Prediksi-prediksi tertentu sering tidak tepat, tetapi kisaran dan variasi penyakit atau keadaan dapat diuraikan. Prognosis sindrom turner adalah baik.5
Komplikasi
11
Pengidap sindrom Turner berisiko tinggi mengalami fraktur semasa kanak-kanan dan osteoporosis pada orang dewasa karena kurangnya estrogen.
Sebagian individu mungkin memperlihatkan ketidakmampuan belajar.
Kelainan imun sering terjadi pada penderita sindroma Turner, termasuk kelainan tiroid (hipotiroid), yang menyebabkan produksi hormon yang mengontrol metabolisme berkurang. Juga dapat terjadi alergi pada gandum sering disebut penyakit Celiac.
Gangguan penglihatan juga dapat terjadi karena fungsi otot mata yang melemah (strabismus) dan tidak dapat melihat jauh.
Penderita sindroma Turner juga sering mengalami gangguan psikologis, seperti percaya diri yang rendah, depresi, kecemasan, kesulitan untuk bersosialisasi, dan gangguan untuk memusatkan perhatian.5
Pencegahan
Pencegahan primer terhadap kelainan genotip memerlukan tindakan sebelum konsepsi. Diagnosis pranatal dengan terminasi kehamilan selektif (pencegahan sekunder) mengubah angka kejadian suatu kelainan. Apabila usaha pencegahan gagal diperlukan suatu tindakan pengobatan.4
Pencegahan primer kelainan genetik
Pada pencegahan, diperlukan peningkatan pengetahuan tentang kedua proses tersebut (kerusakan kromosom). Semua kelainan gen tunggal disebabkan oleh mutasi. Masih diperlukan berbagai penelitian unntuk mencari penyebab kelainan ini. Kelainan yang disebabkan multifaktor mempunyai peranan yang paling besar dalam pencegahan primer. Tujuan disini adalah agar orang yang mempunyai resiko dapat mencegahnya dengan menghindari faktor lingkungan.
Pencegahan sekunder kelainan genetik
Pencegahan sekunder termaksud didalamnya semua aspek uji tapis prenatal dan terminasi selektif. Kelainan kromosom
Uji tapis biokimia untuk menentukan kehamilan resiko tinggi, dalam kombinasi dengan umur ibu, sangat meningkatkan efektifitas program pencegahan pranatal. Biasanya uji tapis dilakukan pada ibu usia 35 tahun keatas dan pada golongan risiko tinggi.
Konseling genetik
Merupakan suatu upaya pemberian advis terhadap orang tua atau keluarga penderita kelainan bawaan yang diduga mempunyai faktor penyebab herediter, tentang apa dan bagaimana kelainan yang dihadapi itu, bagaimana pola penurunannya dan juga upaya untuk melaksanakan pencegahan ataupun menghentikannya. Terdapat tiga aspek konseling:
12
Perkiraan risiko yang seungguhnya
Tindakan suportif untuk memberikan kepastian bahwa pasien dan keluarganya memperoleh manfaat dari nasihat yang diberikan dan tindakan pencegahan yang bisa dilakukan.
Tujuan dari konseling genetik adalah untuk mengumpulkan data-data medis maupun genetik dari pasien ataupun keluarga pasien yang berpotensi dan menjelaskan langkah-langkah yang dapat dilakukan. Konseling genetik dimulai dengan pertanyaan mengenai kemungkinan terjadinya kelainan genetik yang diajukan oleh orang tua/wali penderita. Akan dilakukan pemeriksaan pendukung yang lengkap, untuk mendapatkan diagnosis yang tepat seperti pemeriksaan sitogenetik, analisis DNA, enzim, biokimiawi, radiologi, USG, CT scan, dan sebagainya.5
Kesimpulan
Sindroma Turner adalah kelainan kromosom di mana semua atau bagian dari salah satu kromosom seks tidak ada (manusia tidak terpengaruh memiliki 46 kromosom, dimana 2 adalah kromosom seks). Khas perempuan memiliki 2 kromosom X, tapi dalam sindromTurner, salah satu kromosom seks hilang atau memiliki kelainan lainnya, dimana mahasiswa berusia 21 tahun tersebut dilihat dari ciri khas sindrom turner hipotesis diterima wanita 21 tahun menderita amenore primer ec sindrom turner dan diperlukan pemeriksaan lebih lanjut untuk memastikannya.
Daftar Pustaka
1. Suryo. Genetika manusia. Yogyakarta: Gadjah Mada University Press; 2003.h.247-50. 2. Hull D, Johnshon DI. Dasar-dasar pediatric. Jakarta: EGC; 2008.h.18-9, 228.
3.
Hassan R & Alatas H. Buku kuliah 1 ilmu kesehatan anak. Jakarta: FKUI;2007. h.222. 4. Behrman RE, Kliegman RM, Arvin AM. Ilmu kesehatan anak nelson. Vol 3. Edisi ke-15.Jakarta: EGC; 2000.h.1992-4.
5. Effendi SH, Indrasanto E. Kelainan kongenital. Dalam: Kosim MS, Yunanto A, Dewi R, Sarosa GI, Usman A, penyunting. Buku ajar neonatologi. Edisi ke-1. Jakarta: Badan Penerbit IDAI;2008.h.63-9.
6. Behrman RE, Kliegman RM, Arvin AM. Ilmu kesehatan anak nelson. Vol 1. Edisi ke-15. Jakarta: EGC; 2000.h.396.
7. Yudha EK, wahyuningsih E, Yulianti D, Karyuni PE. Buku saku patofisiologi: genetika. Edisi ke-3. Jakarta: EGC;2007.h.63-6.