Page : 1 of 69
IMPROVEMENT OF ANKLE-BRACHIAL INDEX BY DLBS1033
TREATMENT IN DIABETIC PATIENTS WITH PERIPHERAL
APPROVAL OF FINAL PROTOCOL
Clinical Data Management:
_______________________________________________ __________________
Liana W. Susanto, MBiomed Date
Clinical Project Management:
_______________________________________________ __________________
dr. Prihatini Hendri Date
Director:
_______________________________________________ _________________
Page : 3 of 69
LIST OF KEY STAFF AND RELEVANT DEPARTMENTS
Principal Investigator:
Investigator Team Members:
Study Coordinator
Trial Site:
Clinical Laboratory:
Prof. Dr. dr. Ketut Suastika, SpPD-KEMD
dr. Wira Gotera, SpPD
dr. A. A. Gede Budhitresna, SpPD dr. I Made Pande Dwipayana, SpPD dr. Made Ratna Saraswati, SpPD
dr. Pande Made Oka Arimas, S.Ked
Department of Internal Medicine,
Faculty of Medicine, University of Udayana/Sanglah Hospital Jl. Diponegoro, Denpasar, Bali
Phone: +62 361 227 911 Fax: +62 361 224 206
PT Prodia DiaCRO Laboratories Prodia Tower 3rd Floor
Jl. Kramat Raya No. 150 Jakarta 10430, Indonesia Phone: +6221 3190 3065
Fax: +6221 314 4182 / 3190 5036
Prodia Laboratory Denpasar Jl. Diponegoro No. 192 Denpasar 80114, Indonesia Phone: +62 361 261 001 Fax: +62 361 236 349
Monitors: Dr. Prihatini Hendri Nurul Hidayah, Pharm.
Jalan Boulevard Bintaro Block B7/B1 No. 5 Bintaro Jaya Sector 7
Data Management:
Sponsor:
Liana W. Susanto, MBiomed Fenny, Pharm.
Jalan Boulevard Bintaro Block B7/B1 No. 5 Bintaro Jaya Sector 7
Tangerang 15224 - Indonesia Phone: +62 21 7454 111 Fax: +62 21 7452 623
Jalan Boulevard Bintaro Block B7/B1 No. 5 Bintaro Jaya Sector 7
Page : 5 of 69
TABLE OF CONTENTS
APPROVAL OF FINAL PROTOCOL ... 2
LIST OF KEY STAFF AND RELEVANT DEPARTMENTS ... 3
TABLE OF CONTENTS ... 5
4.3.1 Primary Efficacy Endpoint ... 22
4.3.2 Secondary Efficacy Endpoints ... 22
4.3.3 Other Endpoints ... 22
4.4 SAFETY ENDPOINTS ... 22
4.5 RATIONALE FOR STUDY DESIGN ... 23
4.6 RATIONALE FOR DOSAGE REGIMEN ... 24
5. STUDY SITE AND POPULATION ... 25
5.1 STUDY SITE,PRINCIPAL INVESTIGATOR AND TEAM ... 25
7.1.1 Initial Visit for Run-in Period (Week -2, Initial Screening) – If applicable ... 29
7.1.2 Visit 1 (Week 0, Screening and Baseline) ... 30
7.1.3 Visit 2 (Week 3) ... 31
7.3.1 Ankle-Brachial Index (ABI) ... 34
7.3.2 hs-CRP ... 36
7.3.3 Thromboxane B2 (TXB2) ... 36
7.3.5 D-dimer ... 37
7.5.2 General Clinical Condition and Vital Signs... 38
7.5.3 Height and Body Weight ... 38
7.5.4 Pregnancy Check ... 38
7.5.5 Diabetes Parameters ... 38
7.5.6 Lipid Profile ... 39
7.5.7 Framingham Point Score ... 39
7.6 BLOOD SAMPLE COLLECTION ... 40
8.5 SAMPLING,RANDOMIZATION AND BLINDING PROCEDURE ... 42
8.6 UNBLINDING PROCEDURES ... 42
8.7 CONCOMITANT ILLNESSES AND MEDICATION ... 43
8.7.1 Concomitant Medication during the Trial ... 43
8.7.2 Precautions/Overdosage and Possible Drug Interactions ... 43
9 ADVERSE EVENTS ... 46
10.3 INDEPENDENT ETHICS COMMITTEES/INSTITUTIONAL REVIEW BOARDS... 49
Page : 7 of 69
11 DATA HANDLING AND RECORD KEEPING ... 51
11.1 RULES FOR COMPLETING CRFS ... 51
11.2 CORRECTIONS TO CRFS ... 51
11.3 CRFFLOW ... 51
12 MONITORING AND OVERSIGHT ... 53
13 PREMATURE TERMINATION OF THE TRIAL ... 54
14 CRITICAL DOCUMENTS ... 55
15 RESPONSIBILITIES ... 56
16 DATA MANAGEMENT ... 57
17 EVALUABILITY OF PATIENTS FOR ANALYSIS ... 58
17.1 EFFICACY... 58
17.2 SAFETY ... 58
18 DATA ANALYSIS ... 59
18.1 SAMPLE SIZE CALCULATION... 59
18.2 EFFICACY ANALYSIS ... 59
18.3 SAFETY ANALYSIS... 59
18.4 INTERIM ANALYSIS ... 60
19 REPORTS AND PUBLICATIONS ... 61
20 RETENTION OF CLINICAL TRIAL DOCUMENTATION ... 62
21 INDEMNITY STATEMENT ... 63
22 INVESTIGATOR STATEMENT ... 64
SUMMARY/SYNOPSIS
Objective
1. To investigate clinical efficacy of DLBS1033 in combination with aspirin compared to aspirin alone, for the improvement of ankle-brachial index (ABI) as well as other hemostasis parameters in diabetic patients with peripheral arterial disease.
2. To investigate safety of DLBS1033 in combination with aspirin compared to aspirin alone in diabetic patients with peripheral arterial disease.
Study Design
This is a prospective, randomized, double-blind, double-dummy, and controlled study of DLBS1033 for the improvement of ankle-brachial index in diabetic patients with peripheral arterial disease (PAD). Clinical and laboratory examinations to evaluate the
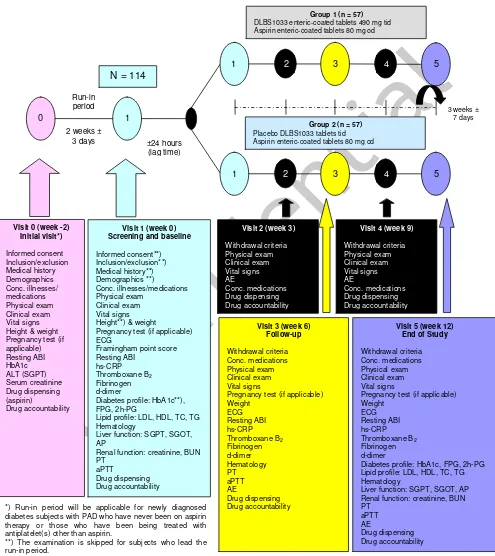
investigational drug’s efficacy and safety will be performed at baseline and at interval of 6 weeks over the 12 week-course of therapy, while drug dispensing and accountability will be carried out at interval of 3 weeks. Study medication will be standard antiplatelet, aspirin at a dose of 80 mg once daily and either the investigational product, DLBS1033 at a dose of 490 mg three times daily or its placebo for 12 weeks. Concomitant medication allowable during study treatment will be any antidiabetes treatment currently used by each individual subjects and lifestyle modification (or intervention). The schematic design of the study is shown in Figure 1.
This study will enrol 114 diabetic patients with PAD, allocated equally in two groups (57 subjects in each group), i.e. The Control Group (will receive aspirin and placebo DLBS1033) and The DLBS Group (will receive aspirin and DLBS1033). In this study, the change (augmentation) of resting ankle-brachial index (ABI) will be measured as the primary efficacy endpoint.
Study Population
Study population will be diabetic patients having PAD with resting ABI of 0.41–0.90.
Key inclusion criteria include:
1. Signed informed consent before any trial related activities. 2. Male or female subjects of 40 – 65 years of age.
3. Diagnosis of diabetes mellitus defined as HbA1c level of 6.5% (for newly diagnosed diabetes) or based on medical history.
4. Presence of peripheral arterial disease with resting ankle-brachial index (ABI) of 0.41–0.90 inclusive (i.e. mild to severe PAD, without critical limb ischemia).
(Note: the reference leg should be determined at this point. Henceforth, ABI improvement will be based on measurement on the same leg as reference).
Page : 9 of 69
6. Patients or patients’ legally acceptable representatives are able and willing to record adverse events in a diary.
7. Patients or patients’ legally acceptable representatives have the ability to comply with the trial protocol.
Key exclusion criteria include:
1. Females of childbearing potential: pregnancy, breast-feeding, and the intention of becoming pregnant.
Patients must accept pregnancy tests during the trial if menstrual cycle is missed.
Fertile patients must use a reliable and effective contraceptive.
2. Recent stroke attack, myocardial infarction/unstable angina/acute coronary syndrome, coronary artery bypass surgery (CABG) or percutaneous transluminal coronary angioplasty (PTCA)/stent within 3 (three) months prior to screening.
3. Impaired liver function: serum ALT > 2.5 times upper limit of normal. 4. Impaired renal function: serum creatinine ≥ 1.5 times upper limit of normal.
5. Concomitant use of other antithrombosis drugs, such as oral anticoagulants, dypiridamole, thienopyridines (such as clopidogrel), cilostazol or glycoprotein IIb/IIIa antagonist or any antiplatelets other than the study medication.
6. Subjects with concurrent herbal (alternative) medicines or food supplements suspected to have effect on hemostasis parameters.
7. Subjects with any other disease state, including chronic or acute systemic infections, uncontrolled illnesses or other chronic diseases, which judged by the investigator, could interfere with trial participation or trial evaluation.
8. Subjects with high risk of bleeding:
Subjects with history of acquired or congenital bleeding disorder, coagulopathy, or platelet disorder;
Subjects with evidence of active pathological bleeding or history of bleeding such as gastrointestinal or genitourinary bleeding unless the cause has been definitely corrected;
History or presence of intracranial bleeding or hemorrhagic retinopathy, unstable angina, severe valvular heart disease, severe anemia, suspected or known dissecting aneurysm, acute myocarditis or pericarditis, severe aortic stenosis, severe hypertrophic obstructive cardiomyopathy,
Severe or uncontrolled arterial hypertension (systolic blood pressure > 160 mmHg, diastolic > 100 mmHg).
9. Subjects with prior experience with DLBS1033 or other oral lumbrokinase products. 10. Subjects with known or suspected allergy to any of study medications used in the
study, including other lumbrokinase products.
11. Subjects with known or suspected allergy or resistant to aspirin.
Withdrawal criteria:
The patient may be withdrawn from the trial at the discretion of the investigator or the sponsor if judged non-compliant with trial procedures or due to safety concerns. A patient must be withdrawn if the following applies:
1. Pregnancy or intention of becoming pregnant.
2. Allergy to any of study medications used in the study.
3. Treatment with any antiplatelets, antithrombosis, anticoagulants, and/or fibrinolytic agents, other than the study medication, including other oral lumbrokinase products, during the course of the study.
4. Any events occurring during the course of the study that might jeopardize the health of the patient.
5. Participation in any other clinical trial during the course of the trial.
Assessments: Efficacy endpoint:
Efficacy endpoint will be measured at baseline, Week 6th of treatment, and at the end of study (Week 12th of treatment).
Primary endpoint:
Change (augmentation) of resting ankle-brachial index (ABI) from baseline to the end of study (week 12th).
Secondary endpoints:
Change of resting ABI from baseline to week 6th of treatment.
Change of hs-CRP from baseline to week 6th and 12th of treatment.
Change of thromboxane B2 (TXB2) from baseline to week 6th and 12th of treatment.
Change of fibrinogen level from baseline to week 6th and 12th of treatment.
Change of d-dimer level from baseline to week 6th and 12th of treatment.
Safety endpoint:
Unless specified time of measurement is mentioned, all safety parameters will be measured at baseline, week 6th, and week 12th of treatment:
Vital signs: blood pressure, heart rate, respiratory rate.
Electrocardiography (ECG).
Routine hematology: Hb, Ht, RBC, WBC, differential of WBC, platelet count.
Liver function:
o Serum ALT (SGPT)
o Serum AST (SGOT)
o Alkaline phosphatase
Liver function will be examined at baseline and at the end of study (week 12th).
Page : 11 of 69 o Serum creatinine
o Blood Urea Nitrogen (BUN)
Renal function will be examined at baseline and at the end of study (week 12th).
Hemostasis parameters:
o Prothrombin Time (PT)
o Activated Partial Thromboplastin Time (aPTT).
Adverse event will be observed and carefully evaluated along the course of the study.
Other Examinations at Screening:
1. Demography data, including: age, sex, weight and height (for BMI calculation), smoking and alcohol status.
2. Interview on medical history, including first-time diagnosis of diabetes (and the type
of DM), family and subject’s history of cardiovascular diseases (coronary arterial disease, peripheral arterial disease, venous thromboembolism, and other atherothrombotic states necessitating medical treatment) or CV events (myocardial infarction, stroke, heart failure).
3. Clinical examination (in addition to laboratory examination) on the presence and degree of the existing microangiopathy complications, such as: retinopathy, neuropathy, and nephropathy.
4. Physical examinations.
5. Vital signs, including blood pressure, heart rate, and respiratory rate. 6. Pregnancy test (for female subjects).
7. Concomitant illnesses and concomitant medications.
8. Diabetes parameters: FPG, HbA1c, and 2-hour plasma glucose.
9. Lipid profile: HDL cholesterol, LDL cholesterol, total cholesterol, and triglycerides. 10. Assessment of Framingham point score.
Trial Products: Investigational drug:
DLBS1033 bioactive protein fraction enteric-coated tablet @ 490 mg
Investigational product will be given one tablet three times daily for twelve weeks.
Standard drug:
Aspirin enteric-coated tablet @ 80 mg (Thrombo Aspilets®, Medifarma) Aspirin will be given one tablet once daily for twelve weeks.
Concomitant medication:
Beta-blockers, ACE inhibitors, calcium antagonists, other blood pressure lowering agents for indication of hypertension, heart failure or any other cardiovascular diseases.
Statins or other cholesterol-lowering agents.
Insulin and/or any oral antidiabetes agents.
Anticoagulant, other fibrinolytic, or antithrombosis agents, any medications other than study medication and permitted concomitant medications, including herbals or food supplements which may affect hemostasis parameters are not allowed during study participation.
Study Allocation and Study Conduct:
There will be two groups of treatment, each consists of 57 subjects receiving either study treatment as follows:
Three times daily Once daily Three times daily
I: treatment group
II: control group
Eligible subjects will be enrolled in the study, randomly allocated into either study group, and instructed to come for study visit at interval of six weeks for efficacy and safety evaluation. Subject should take study medication everyday over twelve weeks of study period.
Diabetic subjects who have been being under therapy with aspirin can directly start with study treatment. For those who are currently not under therapy with aspirin, though, i.e. newly-diagnosed diabetic subjects (with PAD) who have never been on aspirin therapy or those who have been being treated with antiplatelet(s) other than aspirin, there will be a run-in period during which they will receive (or be switched to) aspirin treatment, without any other antiplatelets, for two weeks. After then, they will receive study medication. Such a run-in period with aspirin is set to make a matching or comparable baseline hemostasis condition of all subjects and the period of two weeks is based on the average lifespan of a platelet, which is normally 7–10 days.
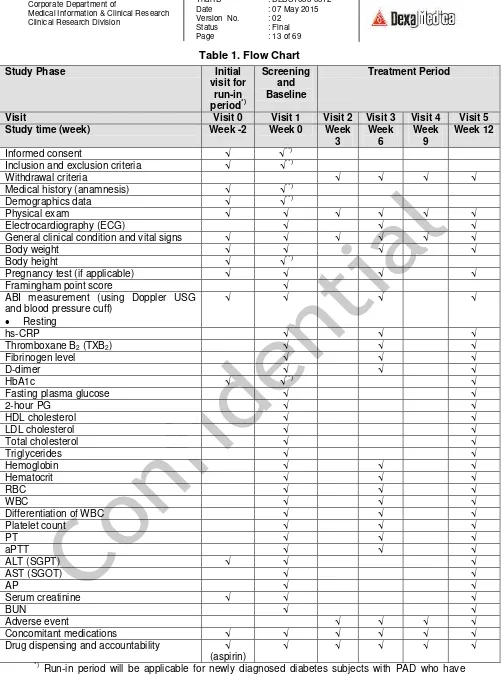
Page : 13 of 69
Inclusion and exclusion criteria √ √**)
Withdrawal criteria √ √ √ √
Medical history (anamnesis) √ √**)
Demographics data √ √**)
Physical exam √ √ √ √ √ √
Electrocardiography (ECG) √ √ √
General clinical condition and vital signs √ √ √ √ √ √
Drug dispensing and accountability √ (aspirin)
√ √ √ √ √
*)
Run-in period will be applicable for newly diagnosed diabetes subjects with PAD who have never been on aspirin therapy or those who have been being treated with antiplatelet(s) other than aspirin. See section 7.1 for further explanation.
1. INTRODUCTION
Peripheral arterial disease (PAD) broadly encompasses the vascular diseases caused primarily by atherosclerosis and thromboembolic pathophysiology processes that alter the normal structure and function of the aorta, its visceral arterial branches, and the arteries of the lower extremity. PAD is the preferred clinical term that should be used to denote stenotic, occlusive, and aneurysmal disease of the aorta and its branch arteries, exclusive of the coronary arteries.1 Patients with PAD may have symptoms, but can also be asymptomatic. The most common symptom of PAD is intermittent claudication, defined as pain, cramping, or aching in the claves, thighs, or buttocks that appears reproducibly with walking exercise and is relieved by rest.2 Patients diagnosed as having PAD, including those who are asymptomatic, have an increased risk of mortality, myocardial infarct, and stroke.3
The major risk factors for PAD are older age (over 40 years), cigarette smoking, and diabetes mellitus. Hyperlipidemia, hypertension, and hyperhomocysteinemia are also important risk factors.4,5,6 Other potential risk factors for PAD include elevated levels of C-reactive protein (CRP), fibrinogen, apolipoprotein B, lipoprotein (a), and plasma viscosity. An inverse relationship has been suggested between PAD and alcohol consumption.2 It is important to note that diabetes is most strongly associated with femoral–popliteal and tibial (below the knee) PAD, whereas other risk factors (e.g. smoking and hypertension) are associated with more proximal disease in the aorto–ilio– femoral vessels.2
In people with diabetes, the risk of PAD is increased by age, duration of diabetes, and presence of peripheral neuropathy. Levels of CRP are also abnormally elevated in patients with impaired glucose tolerance and diabetes; then elevated levels of CRP are strongly associated with the development of PAD.7 Finally, CRP may increase the local production of compounds impairing fibrinolysis, such as plasminogen activator inhibitor (PAI)-1. Studies also showed that PAI-1 levels are elevated in insulin-resistant subjects and associated with increased cardiovascular risk of atherothrombosis.8,9,10 PAI-1 also has positive correlation with body mass index, which is usually high in diabetes patient.10,11 Whereas, evidence shows that in diabetic patients fibrinogen may be a cardiovascular risk factor because it is correlated to increased thrombin formation.12 The existence of both increased fibrinogen and thrombin activation in diabetes has been hypothesized to play a role in the pathogenesis of atherosclerosis in this disease. Therefore, people with diabetes have increased risk of PAD.
Page : 15 of 69
An accurate estimation of the prevalence of PAD in diabetes should rely upon a validated and reproducible test. Such a test is the ankle–brachial index (ABI), which involves measuring the systolic blood pressure in the ankles (dorsalis pedis and posterior tibial arteries) and arms (brachial artery) using a hand-held Doppler and then calculating a ratio. This test has been reported to have a sensitivity above 90% and a specificity of 95% for the diagnosis of peripheral arterial disease.13 Using the ABI, one recent survey found a prevalence of PAD in people with diabetes > 40 years of age to be 20%.14 Moreover, another survey of patients with diabetes > 50 years of age showed a prevalence of PAD of 29%.15 The ABI greater than 1.30 suggesting a non-compressible, calcified vessel. In this condition, the true pressure at that location cannot be obtained and additional tests are required to diagnose PAD. The value of between 0.91–1.30 is considered as normal condition.2,13,16 Patients with leg pain on exertion who have such ABI values should be considered for an exercise test. An ABI value that is over 0.90 at rest, but decreasing by 20% after exercise, is diagnostic of PAD. If the initial ABI value is 0.90 or less at rest, the patient probably has PAD and no additional tests are necessary.17,18
Because of the presence of above-mentioned risk factors, the systemic nature of atherosclerosis and the high risk of ischemic events, diabetic patients with PAD should be considered candidates for secondary-prevention strategies that include aggressive risk-factor modification and antiplatelet drug therapy.2,3,19,20,21 At the present time, it is recommended that patients with PAD be treated with antiplatelet and lipid-lowering agents, along with management of their hypertension, hyperglycemia, and lifestyle modification in order to reduce the risk of cardiovascular complications.
To date, there are three different classes of platelet-inhibiting drugs which are mostly used for the prevention and treatment of atherothrombotic disorders, i.e. cyclooxygenase-1 (COX-1) inhibitors (aspirin), ADP P2Y12 receptor antagonists
Even though aspirin therapy for diabetic patients at increased CVD risk is recommended,20 its benefit to prevent cardiovascular events and death in diabetic patients with PAD is considered equivocal.25,26 There is also a reduced clinical efficacy (aspirin resistance) of aspirin in diabetic compared with a non-diabetic population.19 In CAPRIE study, it was shown that in patients with PAD, clopidogrel was associated with a risk reduction of 24% compared with aspirin. Clopidogrel was also superior to aspirin therapy in PAD subjects who had diabetes. In terms of safety, clopidogrel was as well tolerated as aspirin.27 Based on these results, the Food and Drug Administration (FDA) approved clopidogrel for the reduction of ischemic events in all patients with PAD.2 However, resistance to the agent was also observed in patients with diabetes receiving long-term treatment with clopidogrel, especially in those under insulin therapy.22 Glycoprotein IIb/IIIa inhibitors (abciximab, tirofiban, lamifiban, eptifibatide) are generally used via the parenteral route within the multiple antithrombosis treatment panel for acute coronary syndromes. These agents seem highly effective in diabetes and are recommended in diabetic patients with acute coronary syndromes undergoing percutaneous interventions.22 Unfortunately, these agents failed to show efficacy in oral preparations, making them unsuitable for long-term prevention in a lifelong disease, such as diabetes.
Through relentless effort of searching for a new agent that may benefit diabetic patients with PAD, DLBS1033, a bioactive protein fraction derived from the local earthworm, Lumbricus rubellus, through a patented technology of extraction, was discovered. The earthworm extract possesses eight major proteins with molecular weight below 100 kDa. This specific pattern of proteins possess unique characteristic of DLBS1033, named as LLP (Lumbricus Low molecular weight Proteins). DLBS1033 has been shown to have antithrombosis activities due to its fibrinogen degradation assay, antiplatelet aggregation, and ex vivo antithrombosis assay; and thrombolytic activities demonstrated by fibrin plate assay and clot lysis assay.28 In vitro studies demonstrated reduced expression of several genes involves in inflammatory and atherogenic reaction by DLBS1033, such as NF-B, TNF-, VCAM-1, and P-selectin.29 In the same study, it was shown that DLBS1033 suppressed the expression of MMP-9 gene, a marker of plaque instability, suggesting that this bioactive protein fraction has the ability to control plaque stabilization. Besides, DLBS1033 also down-regulated JAK/STAT1 system which is responsible for vascular smooth muscle cell proliferation.
Page : 17 of 69
Safety profile of DLBS1033 has also been demonstrated in healthy adult subjects.35,36 Based on the evaluation of safety parameters, which included hematology parameters, blood chemistry, urine and stool occult blood evaluation, and ECG interpretation, DLBS1033 was proven safe in healthy subjects. To date, DLBS1033 has been approved by National Agency of Drug and Food Control (NADFC), Republic of Indonesia, to be marketed as Indonesian standardized herbal medicine. Since then, no clinically significant adverse drug reactions have been reported.
2. STUDY RATIONALE
The prevalence of PAD in diabetes appears higher than previously estimated.14,15 People with both PAD and diabetes mellitus are at increased risk of subsequent cardiovascular events.
At the present time, it is recommended that patients with PAD be treated with antiplatelet and lipid-lowering agents, along with management of their hypertension, hyperglycemia, and lifestyle modification in order to reduce the risk of cardiovascular complications. More specific to the antiplatelets and thrombolytic agents, currently available drugs approved by Food and Drug Administration (FDA) have several limitations, such as resistance (to clopidogrel), and parenteral-only dosage forms (of glycoprotein IIb/IIIa inhibitors, such as abciximab, tirofiban, lamifiban, eptifibatide). Even though these agents seem highly effective in diabetes and are recommended in diabetic patients, such limitations have made them unsuitable for long-term prevention in a lifelong disease, such as diabetes. This drives the development of a more effective antiplatelet treatment regimen, particularly for prevention of atherothrombotic events in diabetic patients with PAD, in whom the risk for developing CV events are much higher and the prevalence are rising.
In the last few years, DLBS1033, a bioactive fraction derived from Lumbricus rubellus earthworm, have been developed, possesses both antithrombosis as well as thrombolytic activities.28 Based on its pharmacological activity, safety profile27,28,29,30,31,32,33 as well as its simple administration (i.e. oral preparation), DLBS1033 can be considered as a new potential agent that is hypothetically beneficial for secondary prevention of cardiovascular events, particularly in diabetes mellitus patients. Besides, the thrombolytic activity of DLBS1033 may have a significant role in reducing other potential risk factors related to haemostasis parameters, such as fibrinogen and D-dimer levels.
Page : 19 of 69 3. OBJECTIVES
The objectives of this study are:
1. To investigate clinical efficacy of DLBS1033 in combination with aspirin compared to aspirin alone, for the improvement of ankle-brachial index (ABI) as well as other hemostasis parameters in diabetic patients with peripheral arterial disease.
4. STUDY DESIGN 4.1 Overview
This is a prospective, randomized, double-blind, double-dummy, and controlled study of DLBS1033, over a total of twelve weeks of therapy. There are 96 subjects (48 subjects per arm) planned to complete the study. To anticipate a withdrawal rate of about 15%, a total of 114 subjects are required to be enrolled in the study.
Subjects will be screened consecutively and eligible subjects will be randomized to receive aspirin tablet 80 mg once daily and either the investigational product, DLBS1033 tablet at a dose of 490 mg three times daily, or its placebo. Clinical and laboratory
examinations to evaluate the investigational drug’s efficacy and safety will be performed
at baseline and at the interval of six weeks over the twelve week-course of therapy, while drug dispensing and accountability will be carried out at interval of 3 weeks.
Diabetic subjects who have been being under therapy with aspirin can directly start with study treatment. For those who are currently not under therapy with aspirin, though, i.e. newly-diagnosed diabetic subjects (with PAD) who have never been treated with aspirin, or those who have been being treated with antiplatelet(s) other than aspirin, there will be a run-in period during which they will receive (or be switched to) aspirin treatment, without any other antiplatelets, for two weeks. After then, they will receive study medication. The schematic figure of the study design is shown in Figure 1 (Page 21).
All subjects will also be instructed to follow a lifestyle modification,40 i.e. cease cigarette smoking (for smokers), follow a healthy diet enriched with protein but restricted in saturated fat, cholesterol and salt, and have routine exercise (i.e. aerobic and weight-bearing exercise for at least 4 to 5 times @ 30 minutes, per week) during their participation in the study. There will be no special treatment or control performed by the Investigators concerning the implementation of this lifestyle modification, thus it becomes essential for the Investigators to provide an earnest education and information as well as the reminder to the patient about this matter.
Before its commencement, the study protocol must obtain approval from the Ethics Committee of Medical Faculty, University of Udayana, Bali. Informed consent will be obtained from each clinical trial subject or their legal guardian. Eligible clinical trial subject will receive study medication and be closely monitored by the Investigator during their participation in the study to evaluate the outcome.
4.2 Design and Methods
The study procedures and methods are described schematically in Figure 1 (Page 21). Eligible subjects are randomly allocated into two treatment groups as follow:
1. Treatment group I (DLBS1033)
: DLBS1033 enteric-coated tablets @ 490 mg, three times daily, for 12 weeks
Page : 21 of 69
2. Treatment group II (Control)
: Placebo DLBS1033 enteric-coated tablets, three times daily, for 12 weeks
Aspirin enteric-coated tablet 80 mg, once daily, for 12 weeks
The study medication is provided in double-blind, double-dummy preparations. Subjects take the study medication everyday over twelve weeks of study period and are instructed to come to the clinic for follow-up visits at interval of six weeks.
Figure 1. Scheme of Study Design.
Lipid profile: LDL, HDL, TC, TG Hematology
DLBS1033 enteric-coated tablets 490 mg tid Aspirin enteric-coated tablets 80 mg od
Group 2 (n = 57)
Placebo DLBS1033 tablets tid Aspirin enteric-coated tablets 80 mg od ±24 hours
Diabetes profile: HbA1c, FPG, 2h-PG Lipid profile: LDL, HDL, TC, TG diabetes subjects with PAD who have never been on aspirin therapy or those who have been being treated with antiplatelet(s) other than aspirin.
4.3 Efficacy Endpoints
4.3.1 Primary Efficacy Endpoint
The primary efficacy endpoint will be: change (augmentation) of resting ankle-brachial index (ABI) from baseline to the end of study (week 12th).
4.3.2 Secondary Efficacy Endpoints
The secondary efficacy endpoints are:
Change of resting ABI from baseline to week 6th of treatment.
Change of hs-CRP from baseline to week 6th and 12th of treatment.
Change of thromboxane B2 (TXB2) from baseline to week 6th and 12th of treatment.
Change of fibrinogen level from baseline to week 6th and 12th of treatment.
Change of d-dimer level from baseline to week 6th and 12th of treatment.
4.3.3 Other Endpoints
Other endpoints evaluated which are not directly associated with the efficacy of the investigational product :
Change of HbA1c from baseline to the end of study (week 12th).
Change of fasting plasma glucose (FPG) level from baseline to the end of study (week 12th).
Change of 2-hour post prandial glucose (2-hour PG) level from baseline to the end of study (week 12th).
Change of lipid profile (LDL, HDL, Total Cholesterol, and Triglyceride levels) from baseline to the end of study (week 12th).
4.4 Safety Endpoints
Unless specified time of measurement is mentioned, all safety parameters will be measured at baseline, week 6th, and week 12th of treatment:
Vital signs: blood pressure, heart rate, respiratory rate.
Electrocardiography (ECG).
Routine hematology: Hb, Ht, RBC, WBC, differential of WBC, platelet count.
Liver function:
o Serum ALT (SGPT)
o Serum AST (SGOT)
o Alkaline phosphatase
Liver function will be examined at baseline and at the end of study (week 12th).
Renal function:
o Serum creatinine
o Blood Urea Nitrogen (BUN)
Renal function will be examined at baseline and at the end of study (week 12th).
Page : 23 of 69 o Prothrombin Time (PT)
o Activated Partial Thromboplastin Time (aPTT).
Adverse event will be observed and carefully evaluated along the course of the study.
4.5 Rationale for Study Design
A parallel, randomized, double-blind, double-dummy, and controlled study was considered as the best design to evaluate the efficacy and safety of a new oral fibrinolytic agent, DLBS1033, in combination with aspirin in diabetic patients with PAD. Random allocation will expose every study subject to an equal opportunity to receive either the investigational product (DLBS1033) or its placebo, on top of aspirin treatment. A double-blind design will minimize any possible bias in evaluating the result of the trial. Placebo or dummy tablets of DLBS1033 will be used in this study in order to maintain the double-blinding procedure.
In this study, aspirin alone was chosen as the active control. This is based on the rationale that in line with the investigational drug, DLBS1033, aspirin also exerts its action through the hemostasis system (i.e. as an antiplatelet), and is recommended for prevention of ischemic events in diabetic patients who are at increased CVD risk.20,23,24
To date, we do not have any clinical data yet, comparing the inhibitory effect of DLBS1033 on platelet aggregation to that of aspirin in healthy volunteers. Therefore it is ethically and scientifically considerable to administer the new agent on top of aspirin as the currently recommended standard regimen. Further, such a combination is also expected to produce a synergistic effect as both agents have different mechanism of action in improving vascular system thus preventing cardiovascular events.
In order to set a matching or comparable baseline hemostasis condition in all subjects, before they receive study medication, newly-diagnosed diabetic subjects (with PAD) who have never been on aspirin therapy will be enrolled in a two-week run-in period during which they will be given aspirin; while those who have been being treated with other antiplatelet(s) than aspirin, will be switched to aspirin treatment during the run-in period. Such an aspirin-treated run-in period is set for a period of two weeks based on the average lifespan of a platelet, which is normally 7–10 days.
A three-week interval of follow-up visit was determined to minimize the chance of losing study subjects along the way during the study, as well as to control the compliance of study subjects with the study regimens and procedure. A twelve-week treatment period was regarded appropriate and adequate to examine the augmentation of ankle-brachial index (ABI) as well as all the secondary endpoints after the medication by the investigational products.
In this study, hs-CRP, TXB2, fibrinogen, and d-dimer as risk factors of PAD will also be
and thrombolytic/fibrinolytic activities. Those parameters have been known to have direct or indirect correlation with increasing risk of PAD in diabetes patients.8,9,10,11,12
Meanwhile, safety evaluation of DLBS1033 will also be evaluated in this study, including the evaluation on the vital signs, cardiac, liver and renal function, hematology, hemostasis parameters, as well as other clinical adverse events occurred during the study period, either observed by the investigator or reported by the subjects.
4.6 Rationale for Dosage Regimen
Page : 25 of 69 5. STUDY SITE AND POPULATION
5.1 Study Site, Principal Investigator and Team
Principal Investigator of this study will be Prof. Dr. dr. Ketut Suastika, SpPD-KEMD. In conducting the study, the principal investigator will be assisted by a study coordinator: dr. Pande Made Oka Arimas, S.Ked. and several co-investigators, as follows: dr. Wira Gotera, SpPD, dr. A. A. Gede Budhi Tresna, SpPD, dr. I Made Pande Dwipayana, SpPD, and dr. Made Ratna Saraswati, SpPD.
This will be a single-center-study with the study site at Department of Internal Medicine, Faculty of Medicine, University of Udayana/Sanglah Hospital, situated at Jl. Diponegoro, Denpasar, Bali.
Clinical laboratory assigned for this study will be Prodia Laboratory Denpasar, situated at Jl. Diponegoro No. 192, Denpasar 80114, Indonesia.
5.2 Number of Patients to be Studied
Planned number of enrolled subjects: 114 subjects (57 subjects in each group) Anticipated withdrawal rate during the study period: 15%
Planned number of complete subjects: 96 subjects (48 subjects in each group)
5.3 Patient Replacement
There will be no patient replacement in the study for every withdrawn subject.
5.4 Nature of Population
Study population will be diabetes patients with PAD with resting ABI of 0.41–0.90 who attend the study site.
5.5 Subject Eligibility 5.5.1 Inclusion Criteria
1. Signed informed consent before any trial related activities. 2. Male or female subjects of 40 – 65 years of age.
3. Diagnosis of diabetes mellitus defined as HbA1c level of 6.5% (for newly diagnosed diabetes) or based on medical history.
4. Presence of peripheral arterial disease with resting ankle-brachial index (ABI) of 0.41–0.90 inclusive (i.e. mild to severe PAD, without critical limb ischemia).2,16,39 (Note: the reference leg should be determined at this point. Henceforth, ABI improvement will be based on measurement on the same leg as reference).
5. Able to take oral medication.
6. Patients or patients’ legally acceptable representatives are able and willing to record adverse events in a diary.
5.5.2 Exclusion Criteria
1. Females of childbearing potential: pregnancy, breast-feeding, and the intention of becoming pregnant.
Patients must accept pregnancy tests during the trial if menstrual cycle is missed.
Fertile patients must use a reliable and effective contraceptive.
2. Recent stroke attack, myocardial infarction/unstable angina/acute coronary syndrome, coronary artery bypass surgery (CABG) or percutaneous transluminal coronary angioplasty (PTCA)/stent within 3 (three) months prior to screening.
3. Impaired liver function: serum ALT > 2.5 times upper limit of normal. 4. Impaired renal function: serum creatinine ≥ 1.5 times upper limit of normal.
5. Concomitant use of other antithrombosis drugs, such as oral anticoagulants, dypiridamole, thienopyridines (such as clopidogrel), cilostazol or glycoprotein IIb/IIIa antagonist or any antiplatelets other than the study medication.
6. Subjects with concurrent herbal (alternative) medicines or food supplements suspected to have effect on hemostasis parameters.
7. Subjects with any other disease state, including chronic or acute systemic infections, uncontrolled illnesses or other chronic diseases, which judged by the investigator, could interfere with trial participation or trial evaluation.
8. Subjects with high risk of bleeding:
Subjects with history of acquired or congenital bleeding disorder, coagulopathy, or platelet disorder;
Subjects with evidence of active pathological bleeding or history of bleeding such as gastrointestinal or genitourinary bleeding unless the cause has been definitely corrected;
History or presence of intracranial bleeding or hemorrhagic retinopathy, unstable angina, severe valvular heart disease, severe anemia, suspected or known dissecting aneurysm, acute myocarditis or pericarditis, severe aortic stenosis, severe hypertrophic obstructive cardiomyopathy,
Severe or uncontrolled arterial hypertension (systolic blood pressure > 160 mmHg, diastolic > 100 mmHg).
9. Subjects with prior experience with DLBS1033 or other oral lumbrokinase products. 10. Subjects with known or suspected allergy to any of study medications used in the
study, including other lumbrokinase products.
11. Subjects with known or suspected allergy or resistant to aspirin.
Page : 27 of 69 5.5.3 Withdrawal Criteria
The patient may be withdrawn from the trial at the discretion of the investigator or the sponsor if judged non-compliant with trial procedures or due to safety concerns. A patient must be withdrawn if the following applies:
1. Pregnancy or intention of becoming pregnant.
2. Allergy to any of study medications used in the study.
3. Treatment with any antiplatelets, antithrombosis, anticoagulants, and/or fibrinolytic agents, other than the study medication, including other oral lumbrokinase products, during the course of the study.
4. Any events occurring during the course of the study that might jeopardize the health of the patient.
5. Participation in any other clinical trial during the course of the trial.
5.6 Rationale for the Inclusion/Exclusion Criteria
Since the study aims to evaluate further the efficacy of DLBS1033 for the improvement of ankle-brachial index in diabetic patients with peripheral arterial, it is regarded appropriate to involve diabetes patients with the presence of PAD, shown by resting ABI of 0.41–0.90. In this study, the diagnosis of diabetes will be defined by either HbA1c level at Screening (for those of newly diagnosed diabetes mellitus) or based on medical history (for those with previously known diagnosis). Current treatment taken by the subject may also be supportive for the confirmation of the diagnosis. In the study, HbA1c criterion is not the only one we use to define diagnosis of diabetes mellitus since some or even most type-2-diabetes mellitus patients with good glycemic control may have HbA1c as well as FPG levels within normal range.
Despite the preclinical as well as clinical evidence that the investigational product does not affect liver and renal function, subjects with inadequate liver and renal function will be excluded for the sake of subjects’ safety, since the study is reckoned as the first clinical study in diabetic subjects.
Since the investigational product has antithrombosis as well as fibrinolytic activities, and in the study there will be a group receiving both the investigational product and aspirin, thus subjects with high-risk of bleeding will be excluded.
6. STUDY SCHEDULE
Planned inclusion of first patient (FPFV): June 2015
Planned recruitment period: 12 months
Planned trial period per subject: 3 months
Planned inclusion of last patient (LPFV): June 2016
Planned completion of the last patient (LPLV): September 2016
Page : 29 of 69 7. STUDY ASSESSMENT – PLAN AND METHODS 7.1 Visit Schedule
Subjects will attend the clinic at:
Initial Visit (Week -2) : Initial Screening for Run-in Period
Visit 1 (Week 0) : Screening and Baseline, 2 weeks ± 3 days after Initial Visit Visit 2 (Week 3) : 3 weeks ± 7 days after Visit 1
Visit 3 (Week 6) : 3 weeks 7 days after Visit 2 Visit 4 (Week 9) : 3 weeks ± 7 days after Visit 3 Visit 5 (Week 12) : 3 weeks 7 days after Visit 4
7.1.1 Initial Visit for Run-in Period (Week -2, Initial Screening) – If applicable
This visit will only be applicable to subjects who have to lead a run-in period, i.e. newly diagnosed diabetes subjects with PAD who have never been on aspirin therapy or those who have been being treated with antiplatelet(s) other than aspirin. Otherwise, please skip this Visit and go directly to Visit 1 (Screening & Baseline).
Subjects will be asked to attend a Screening Visit whereby they will be informed thoroughly about the study and their rights while participating in the study. Signed Informed Consents must be obtained prior to any trial related activities which is defined as any procedure that would not have been performed during normal management of the patient. All patients who have given their informed consent will be screened against the inclusion and exclusion criteria and recorded in the Case Report Forms (CRFs).
The following procedures will also be performed to ensure their eligibility:
Demographic information.
Medical history, including history of drug or alcohol abuse.
Physical examination.
General clinical condition.
Vital signs, including: blood pressure, heart rate, and respiratory rate.
Body height and weight.
Pregnancy test (if applicable).
Ankle-brachial index measurement (resting).
Blood sampling for measurement of:
o Diabetes parameters: HbA1c
o Liver function (SGPT)
o Renal function (serum creatinine)
Concomitant illness and concomitant medication.
recorded and listed in the Screening Log. The Investigator must keep a log identifying all patients having signed informed consent.
To each of eligible patients, the following must be carried out:
Aspirin tablets sufficient for the run-in period (2 weeks) will be provided to the patient and recorded in the Drug Accountability Form.
A patient diary will be given to the patient to record any adverse events and concomitant medication during the run-in period.
All patients will be instructed to bring the Diary and the leftover study medication, including the bottle packs, at the next visit.
An appointment for Visit 1 will be made 2 weeks 3 days after this Initial Visit.
7.1.2 Visit 1 (Week 0, Screening and Baseline)
For subjects who lead the run-in period, they will not be asked again for their written consent to participate in the study. The Procedure A will be skipped and ProcedureB
will be followed.
For subjects who do not lead the run-in period, they will be asked to attend a Screening Visit whereby they will be informed thoroughly about the study and their rights while participating in the study. Signed Informed Consents must be obtained prior to any trial related activities. All patients who have given their informed consent will be screened against the inclusion and exclusion criteria and recorded in the Case Report Forms (CRFs). They will follow both Procedure A and Procedure B.
The following procedures will be performed to ensure subjects’ eligibility:
Procedure A.
Demographic information.
Medical history, including history of drug or alcohol abuse.
Body height.
Blood sampling for measurement of HbA1c.
Procedure B.
Physical examination.
General clinical condition.
Vital signs, including: blood pressure, heart rate, and respiratory rate.
Pregnancy test (if applicable).
Electrocardiography (ECG).
Body weight.
Pregnancy test (if applicable).
Assessment of Framingham point score.
Ankle-brachial index measurement (resting).
Page : 31 of 69 o Glycemic control parameters: FPG, 2-hour PG
o Lipid profile: HDL, LDL, total cholesterol, triglycerides
o Inflammatory marker: hs-CRP
o Hemostasis parameters: thromboxane B2 (TXB2), fibrinogen, d-dimer, PT, aPTT o Routine hematology: hemoglobin, hematocrit, RBC, WBC, differentiation of
WBC, platelet count
o Liver function: SGPT, SGOT, AP
o Renal function: serum creatinine, BUN. Concomitant illness and concomitant medication.
When all data relating to screening visit are obtained, the data must be reviewed by the investigator to ensure that the patient is eligible for the trial. If the patient is not eligible, the patient is a Screening Failure. The reason for exclusion of screening failures will be recorded and listed in the Screening Log. The Investigator must keep a log identifying all patients having signed informed consent.
To each of eligible patients, the following must be carried out:
A subject ID peculiar to each study subject will be given.
Study medication sufficient for the period from Visit 1 to Visit 2 will be provided to the patient and recorded in the Drug Accountability Form.
A patient diary will be given to the patient to record any adverse events and concomitant medication from the day of Visit 1 onwards.
All patients will be instructed to bring the Diary and the leftover study medication, including the bottle packs, at the next visit.
An appointment for Visit 2 will be made 3 weeks 7 days after visit 1.
7.1.3 Visit 2 (Week 3)
Subjects will attend the clinic for a follow-up visit. The following will be performed by the Investigator on the study subjects and all relevant information will be recorded in the CRFs:
Evaluation of trial continuation according to withdrawal criteria.
Physical examination.
General clinical condition.
Vital signs, including: blood pressure, heart rate, and respiratory rate.
Recording of changes in concomitant medication.
Recording of adverse event since last visit. Any worsening in concomitant illness or new illness must be recorded as an adverse event.
Collection of the leftover trial product from previous visit brought by each patient and recording it in the Drug Accountability Form.
The previous visit patient diary will be collected and the subject will be given another new one to record any adverse event and concomitant medication during each period.
All subjects will be instructed to bring the Diary and the leftover trial medication (including the bottle packs) at the next visit.
An appointment for next visit will be made 3 weeks 7 days after current visit.
Between current visit and the next visit, the patients can contact the trial doctor (investigator) in case of any medical emergency or if needed.
7.1.4 Visit 3 (Week 6)
Subjects will attend the clinic for a follow-up visit. The following will be performed by the Investigator on the study subjects and all relevant information will be recorded in the CRFs:
Evaluation of trial continuation according to withdrawal criteria.
Physical examination.
General clinical condition.
Vital signs, including: blood pressure, heart rate, and respiratory rate.
Pregnancy test (if applicable).
Electrocardiography (ECG).
Body weight.
Ankle-brachial index measurement (resting).
Blood sampling for measurement of:
o Glycemic control parameters: FPG, HbA1c, 2-hour PG
o Lipid profile: HDL, LDL, total cholesterol, triglycerides
o Inflammatory marker: hs-CRP
o Hemostasis parameters: TXB2, fibrinogen, d-dimer, PT, aPTT
o Routine hematology: hemoglobin, hematocrit, RBC, WBC, differentiation of
WBC, platelet count
o Liver function: SGPT, SGOT, AP
o Renal function: serum creatinine, BUN. Recording of changes in concomitant medication.
Recording of adverse event since last visit. Any worsening in concomitant illness or new illness must be recorded as an adverse event.
Collection of the leftover trial product from previous visit brought by each patient and recording it in the Drug Accountability Form.
Study medication sufficient for the period from current visit to the next one will be provided to the patient and recorded in the Drug Accountability Form.
Page : 33 of 69
All subjects will be instructed to bring the Diary and the leftover trial medication (including the bottle packs) at the next visit.
An appointment for next visit will be made 3 weeks 7 days after current visit.
Between current visit and the next visit, the patients can contact the trial doctor (investigator) in case of any medical emergency or if needed.
7.1.5 Visit 4 (Week 9)
Subjects will attend the clinic for a follow-up visit. The following will be performed by the Investigator on the study subjects and all relevant information will be recorded in the CRFs:
Evaluation of trial continuation according to withdrawal criteria.
Physical examination.
General clinical condition.
Vital signs, including: blood pressure, heart rate, and respiratory rate.
Body weight.
Recording of changes in concomitant medication.
Recording of adverse event since last visit. Any worsening in concomitant illness or new illness must be recorded as an adverse event.
Collection of the leftover trial product from previous visit brought by each patient and recording it in the Drug Accountability Form.
Study medication sufficient for the period from current visit to the next one will be provided to the patient and recorded in the Drug Accountability Form.
The previous visit patient diary will be collected another new one will be given to record any adverse event and concomitant medication during each period.
All subjects will be instructed to bring the Diary and the leftover trial medication (including the bottle packs) at the next visit.
An appointment for next visit will be made 3 weeks 7 days after current visit.
Between current visit and the next visit, the patients can contact the trial doctor (investigator) in case of any medical emergency or if needed.
7.1.6 Visit 5 (Week 12, End of Study)
Subjects will attend the clinic in the morning after an overnight fast. The following will be performed and all relevant information will be recorded in the CRFs:
Evaluation of trial continuation according to withdrawal criteria.
Physical examination.
General clinical condition.
Vital signs, including: blood pressure, heart rate, and respiratory rate.
Pregnancy test (if applicable).
Electrocardiography (ECG).
Ankle-brachial index measurement using Doppler USG and blood pressure cuff (resting).
Blood sampling for measurement of:
o Glycemic control parameters: FPG, HbA1c, 2-hour PG
o Lipid profile: HDL, LDL, total cholesterol, triglycerides
o Inflammatory marker: hs-CRP
o Hemostasis parameters: TXB2, fibrinogen, d-dimer, PT, aPTT
o Routine hematology: hemoglobin, hematocrit, RBC, WBC, differentiation of
WBC, platelet count
o Liver function: SGPT, SGOT, AP
o Renal function: serum creatinine, BUN. Recording of changes in concomitant medication.
Besides, the following will also be performed:
Recording of adverse events since last visit. Any worsening in concomitant illness or new illness must be recorded as an adverse event.
Collection of patient diary(ies).
Collection of the leftover trial product from previous visit(s) brought by each patient and recording it in the Drug Accountability Form.
Completion of End of Study Form.
7.2 Premature Discontinuation of Patient’s Participation
In case of any premature discontinuation of the study products, the patient will, if possible, be called in for a last visit within 7 days after study products discontinuation. That last visit will be necessary particularly for subjects who have been on the study medication for at least 14 days. At the end of study visit, clinical safety evaluation should be performed for subjects who have taken at least one dose of study medication. Even if the patient is not able to attend, the End of Trial Form must be completed and the Drug Accountability Form must be filled in.
7.3 Assessment for Efficacy 7.3.1 Ankle-Brachial Index (ABI)
ABI has been widely adopted for confirmation of a clinical diagnosis of peripheral arterial disease and its quantification.13 Simple to perform; it is a non-invasive, quantitative measurement of the patency of the lower extremity arterial system. Compared with an assessment of pulses or a medical history, ABI has been found to be more accurate.2
The few contraindications for the measurement of the ABI include excruciating pain in
Page : 35 of 69
incompressible (such as in elderly patients, patients with diabetes, or patients with end-stage renal failure requiring dialysis), these conditions are not absolute contraindications to measuring the ABI.41
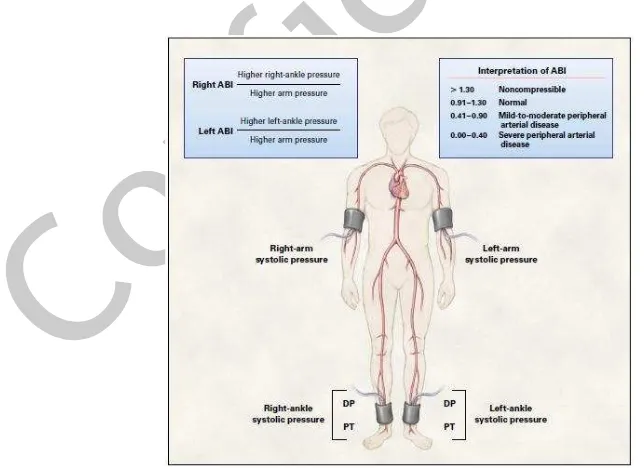
To measure an ABI, the following equipments are needed: a continuous-wave Doppler machine, ultrasonic gel, and a sphygmomanometer with a blood-pressure cuff. Systolic blood pressure is measured by Doppler ultrasonography in each arm and in the dorsalis pedis (DP) and posterior tibial (PT) arteries in each ankle. To calculate the ABI, divide the systolic blood pressure in the ankle by the systolic blood pressure in the arm. The higher brachial systolic pressure is usually chosen for calculation, simply because the vessels of an arm may be affected by arterial occlusive disease. The higher of the systolic pressures from the dorsalis pedis or posterior tibial artery is used to determine the ABI.41
With a patient at rest, a normal ABI ranges from 0.91 to 1.30. A reading above 1.30 is usually suggestive of incompressible tibial arteries. Decreases in the ABI are consistent with peripheral arterial disease. Mild-to-moderate peripheral arterial disease usually produces an ABI in the range of 0.41 to 0.90. A reading below 0.40 suggests the presence of severe peripheral arterial disease.17,42
The ABI commonly measured at rest, even though ABI measurements coupled with exercise testing may enhance the sensitivity of the test and may identify additional patients with PAD who have normal resting ABI values.17,43 Resting ABI values are independently associated with late overall mortality and cardiac death.44
Figure 2. Measurement of the Ankle-Brachial Index (ABI)
Baseline. If both legs show PAD, however, the ABI of each leg will still be measured and recorded. The ABI will be measured by Investigator at study site at initial screening, screening/baseline, week 6th, and at the end of study (week 12th).
7.3.2 hs-CRP
CRP is an acute-phase protein, which is released in the circulation in response to inflammation and tissue damage. Regardless of various hs-CRP methods being used, the CRP molecule measured is the same. Hs-CRP level less than 1 mg/L are
considered “low”, levels from 1 to 3 mg/L are considered “average”, and levels greater than 3 mg/L are considered “high”. Because CRP can fluctuate over time, most experts
recommend measuring two hs-CRP levels a few weeks apart and average the two values.45
A strong predictive relationship between hs-CRP levels and future atherothrombotic events, one of which peripheral arterial disease, has been shown by several studies. It is said that hsCRP levels were associated with major CVD risk factors, 10-year CVD risk, lower ABI, and higher CIMT values. The measurement of hs-CRP will be performed at screening/baseline, week 6th, and at the end of study (week 12th) by the assigned Clinical Laboratory.
7.3.3 Thromboxane B2 (TXB2)
Thromboxane A2 (TXA2) is a cyclooxygenase product of arachidonic acid that causes
irreversible platelet aggregation. Biochemical assessment of TXA2 biosynthesis would
complement the use of pharmacological agents to elucidate the role of this compound in the mediation of human disease. TXA2 is extremely evanescent in biological fluids and
cannot be measured directly. Thus, attention has consequently focused on the measurement of more stable, but biologically inactive, metabolites. The analysis of thromboxane B2 (TXB2), the metabolite of TXA2 hydrolysis in plasma, has been widely
employed to assess the formation of TXA2 by platelets in the circulation. 46
The quantitative determination of TXB2 concentrations will be performed by the assigned
Clinical Laboratory at screening/baseline, week 6th, and at the end of study (week 12th).
7.3.4 Fibrinogen level
Page : 37 of 69 7.3.5 D-dimer
According to the study conducted by McDermott et al.,48 higher level of inflammation marker, d-dimer, is associated with characteristics in persons with PAD. D-dimer as final product of fibrinolysis may elevate the inflammatory cascade by activating neutrophils and monocytes. D-dimer will be measured at screening/baseline, week 6th, and at the end of study (week 12th).
7.4 Assessment for Safety 7.4.1 Routine Hematology
Hematological examinations which include measurement of hemoglobin level, hematocrit, red blood cells (RBC), white blood cells (WBC), differentiated cells of WBC, as well as platelet count will be performed by the assigned Clinical Laboratory at screening/baseline, week 6th, and at the end of study (week 12th).
7.4.2 Liver Function
Measurement of liver function will be performed by the assigned Clinical Laboratory at screening/baseline and at the end of study (week 12th). The measurement will consist of:
Serum ALT (SGPT)
Serum AST (SGOT)
Alkaline phosphatase (AP)
In addition, serum ALT will also be measured at the initial screening.
7.4.3 Renal Function
Measurement of renal function will be performed by the assigned Clinical Laboratory at screening/baseline and at the end of study (week 12th). The measurement will consist of:
Serum creatinine
BUN
In addition, serum creatinine will also be measured at the initial screening.
7.4.4 Electrocardiography (ECG)
7.4.5 Haemostasis Parameters
Two haemostasis parameters, prothrombin time (PT) and activated partial thromboplastin time (aPTT) measurement will be performed by the assigned Clinical Laboratory at screening/baseline, week 6th, and at the end of study (week 12th).
7.4.6 Adverse Event
Any adverse event occur during the subjects participation in the study (including ones which are spontaneously reported by the subjects if observed by the investigator or recorded in patient diary) will be recorded at each visit according to the procedures described in Section 9. Adverse Event.
7.5 Other Assessment 7.5.1 Physical Examination
A general physical examination will be performed at study site, at each study visit, including cardiovascular system, respiratory system, musculoskeletal system, nervous system, abdomen, and extremities.
7.5.2 General Clinical Condition and Vital Signs
General clinical condition and vital signs will be measured at study site, at each study visit. Vital signs include blood pressure, heart rate, and respiratory rate.
7.5.3 Height and Body Weight
For each patient, the height (without taking the shoes’ height into account) will be recorded at screening/baseline visit, whereas weight at screening/baseline, week 6th, and week 12th. The body mass index (BMI) for each patient at such individual visits will be calculated from the data.
7.5.4 Pregnancy Check
Pregnancy test may not be necessarily performed. However, for female subjects of childbearing potential, Investigator should make sure at screening and at any time during the trial that the subjects are not conceived. They should also use non-hormonal contraception during their study participation.
7.5.5 Diabetes Parameters
Page : 39 of 69 7.5.6 Lipid Profile
In order to evaluate the metabolic control of study subjects, which is actually not directly associated with the efficacy of the investigational product, lipid profile consisting of total cholesterol, LDL cholesterol, HDL cholesterol, and triglyceride levels will be measured at screening/baseline and at the end of study (week 12th) by the assigned Clinical Laboratory.
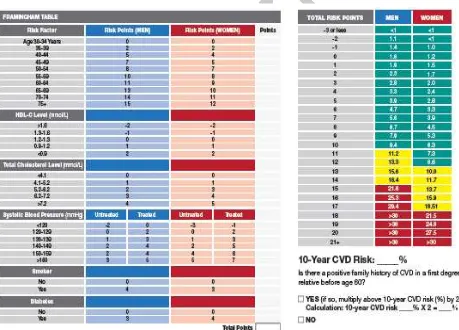
7.5.7 Framingham Point Score49
Clinical strategies for prevention of new-onset coronary heart disease (CHD) are often based on the prediction of 10-year risks. With the third report of the National Cholesterol Education Program, an updated risk prediction system, the Framingham Point Scores (FPS), was presented and recommended. The FPS is represented in the Adult Treatment Panel (ATP) III as risk charts separate for men and women. Points assigned to categorized risk factors are summed overall risk factors, and the summary score corresponds to a 10-year predicted risk of CHD. The assessment of FPS will be performed at screeening/baseline according to the FPS as shown in Figure 3.49
7.6 Blood Sample Collection
All blood samples used in the study will be stored properly at least until all data analysis has been completed and the final study report issued. Before discarding the samples, consent has to be obtained by the Clinical Laboratory from Dexa Medica. The responsibility to keep retaining the blood samples till a period of time will also be specified in a separate, written agreement between Dexa Medica and the assigned Clinical Laboratory.
7.7 Patient Compliance
The investigator will remind the patient to follow the procedures describes in the protocol at each visit, including study dose regimen, visit procedures and recording any adverse events and concomitant medication in the patient diary.
7.8 Protocol Waivers and Violation
Deviation (waivers and violation) from the protocol should not occur. If deviation occurs, the Investigator must inform the monitor, and the implication of the deviation must be reviewed and discussed. Any deviation must be documented (recorded in Note to File Form), starting the reason and date, the action taken, and the impact for the patient and/or the trial. The documentation must be kept in the Investigator’s trial File and the