Factor? Implications for Cholinergic Therapies in
Alzheimer’s Disease
Marcus Rattray
Studies on the neurobiology of nerve growth factor (NGF) reveal a diverse range of actions. Through alterations in gene expression, NGF is important in maintaining and regulating the phenotype of neurons that express the high-affinity receptor, trkA. Nerve growth factor also has a rapid action, revealed by its role in pain signaling in bladder and in skin. In the central nervous system (CNS), NGF has an intimate relationship with the cholinergic system. It promotes cholinergic neuron survival after experimental injury but also maintains and regulates the phenotype of uninjured cholinergic neurons. In addition to these effects mediated by gene expression, NGF has a rapid neurotransmitter-like action to regulate cholinergic neurotransmission and neuronal excitability. Consistent with its actions on the cholinergic system, NGF can enhance function in animals with cholinergic lesions and has been proposed to be useful in humans with Alzhei-mer’s disease (AD); however, the problems of CNS deliv-ery and of side effects (particularly pain) limit the clinical efficacy of NGF. Drug treatment strategies to enhance production of NGF in the CNS may be useful in the treatment of AD. Nicotine is one such agent, which, when administered directly to the hippocampus in rats, produces long-lasting elevation of NGF production. Biol Psychi-atry 2001;49:185–193 © 2001 Society of Biological Psychiatry
Key Words: Neurotrophin, hippocampus, acetylcholine, glutamate, NGF, trkA
Introduction
N
erve growth factor (NGF) is the prototypic neurotro-phic factor. In humans and other mammals, the neurotrophins comprise a family of four similar proteins named NGF, brain-derived neurotrophic factor (BDNF),neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5) (Lindsay et al 1994). The first member of this family to be characterized was NGF, and it remains the best studied. This molecule exists as a dimer of two identical polypep-tide chains, each of 118 amino acid residues (McDonald and Blundell 1991). All neurotrophins have a similar overall structure, with a small number of amino acid differences between the neurotrophins determining the specificity of receptor binding.
The best understood neurotrophin receptors are the family of proteins known as tyrosine receptor kinase (trk). Within this family are three distinct but closely related receptors known as trkA (p140trk), trkB, and trkC. Each trk receptor subunit crosses the cell membrane a single time. The extracellular portion of each trk forms the neurotro-phin-binding site, with differences in amino acid residues determining binding selectivity. NGF binds to trkA, BDNF, and NT-4/5 bind to trkB. Neurotrophin-3 binds relatively selectively to trkC but also can bind to trkA and trkB (Ultrech et al 1999). The intracellular portion of the receptor contains a tyrosine kinase (TK) domain and various sites that can dock with other proteins.
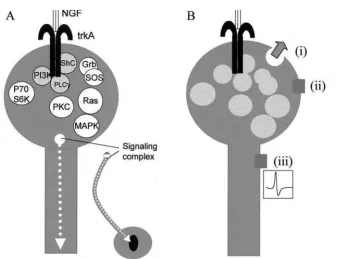
Binding of NGF to trkA causes receptor dimerization, enabling an active complex of two trk receptor subunits with the neurotrophin molecules to be formed. This change of shape results in phosphorylation of three ty-rosine residues on each subunit within the TK domains. The activated TK domains catalyze the phosphorylation of several adjacent tyrosine residues within the intracellular regions of each receptor subunit. These phosphorylated regions are then able to dock with, and activate by phosphorylation, a number of enzymes and proteins in the nerve ending (Figure 1A). Docked proteins bind to addi-tional proteins and activate them (Figure 1A). Thus, a large number of signaling proteins become recruited and activated as a consequence of NGF binding to the trk receptor. These include phospholipase C gamma, which mediates phospholipid hydrolysis, and a number of protein kinases (including PI3 kinase, S6 kinase, protein kinase C, and mitogen-activated protein [MAP] kinase), which are capable of phosphorylating a wide range of target proteins.
From the Biochemical Neuropharmacology Group, Centre for Neuroscience Re-search, GKT School of Biomedical Sciences, King’s College London, London, United Kingdom.
Address reprint requests to Marcus Rattray, GKT School of Biomedical Sciences, King’s College London, Centre for Neuroscience Research, Hodgkin Building, Guy’s Hospital Campus, London SE1 1UL, UK.
Received May 23, 2000; revised August 14, 2000; accepted August 22, 2000.
© 2001 Society of Biological Psychiatry 0006-3223/01/$20.00
The complex cascade of intracellular signaling events underlies the biological effects of the neurotrophins (Friedman and Greene 1999).
The complex of NGF/trkA with docked proteins, in-cluding phospholipase C gamma, is endocytosed at the synapse to form signaling complexes that are retrogradely transported along the axon to the cell body (Grimes et al 1996). When the signaling complex reaches the neuronal cell body, it will phosphorylate transcription factors, including cAMP response element-binding protein (CREB), which can then enter the nucleus (Riccio et al 1997). In this manner, trk receptor activation is directly linked to alteration of gene expression. Alterations in the pattern of gene expression underlie the maintenance, survival, and plasticity effects of NGF and other neuro-trophins on their target neurons. In addition to these events, which would be predicted to occur hours to days after neurotrophin binding to trk receptors, there is an increased awareness that NGF and other neurotrophins can exert more rapid effects on neurotransmitter synthesis, release, and the electrical activity of neurons by altering the activity of proteins involved in synaptic transmission (Figure 1B).
Another neurotrophin receptor exists, named p75NTR, a member of the TNF receptor/Fas/CD40 superfamily. p75NTR, binds to NGF and other neurotrophins with approximately equal affinity (Chao 1994). Receptor acti-vation stimulates several intracellular events that are quite distinct to those mediated by trk receptors. The p75NTR receptor does not contain tyrosine kinase activity but instead mediates several distinct biochemical effects in-cluding sphingolipid hydrolysis and activation of the c-Jun amino terminal protein kinase (JNK) signaling pathway (Friedman and Greene 1999).
Current evidence suggests that for cells that express p75NTR but not a trk receptor, the dominant effect of neurotrophin-mediated p75NTR signaling is cell death. Neurotrophin signaling through p75NTR plays an impor-tant role in programmed cell death during the development of the nervous system (Friedman and Greene 1999). In some cholinergic neurons of the central nervous system (CNS), and in all neurotrophin-sensitive sensory and sympathetic neurons in the peripheral nervous system, p75NTRis coexpressed with one or more trk receptors. For these neurons, the death signals from p75NTRactivation are suppressed by interactions with trk or signals from trk.
Figure 1. Nerve growth factor (NGF) signaling at the synapse.(A)At the nerve terminal, NGF binds to the trkA receptor. Activation of the receptor complex stimulates docking of a number of proteins; shown here are phosphatidyl inositol 3 kinase (PI3K), ShC, and phospholipase Cg(PLCg). ShC activates the SOS/Grb complex to activate the small GTPase, Ras, and MAP kinase (MAPK). Other
In this case, cell death does not occur in response to neurotrophins; instead p75NTRsignaling somehow
modu-lates the effect of trk receptor activation to suppress the death signal.
Models for NGF Effects: Lessons from the
Peripheral Nervous System
Until recently, our knowledge of the functions of NGF and the other neurotrophins has arisen from studies of the developing nervous system. The neurotrophic concept states that the tissue to be innervated by a particular group of neurons produces a limiting amount of a trophic factor that, during a critical stage in target innervation, binds to receptors, is internalized, and retrogradely transported to the cell bodies of these neurons. This supply of neurotro-phin is critical to the development of an appropriate phenotype and connectivity of the target neuron. For developing neurons at least, a supply of the appropriate neurotrophins is essential for cell survival. These concepts have been extensively tested and refined, particularly in the peripheral nervous system.
In the adult, neurotrophin expression continues to be widespread, suggesting a role of these factors beyond that of neuronal development. For example, NGF is produced by many peripheral tissues, in particular, the skin and other tissues that receive a rich sensory or sympathetic innervation. Many sympathetic neurons in the adult ex-press trkA (and p75NTR) receptors, as do a class of primary sensory neurons that are particularly important in acute and inflammatory pain (McMahon 1996; Snider and Mc-Mahon 1998). Although it is clear that subgroups of sensory neurons retain their sensitivity to neurotrophins, it is also clear that a supply of neurotrophins is not necessary for the survival of these responsive neurons. Therefore, a new appreciation of the roles of these factors is emerging that does not fit with the classical neurotrophic concept: NGF is now known to play a role in cell survival after injury, regeneration, collateral sprouting, neuronal size and complexity, expression of transmitters, and neuronal sensitivity (McMahon et al 1995; McMahon 1996; Woolf 1996).
In the context of the adult nervous system, NGF is the best studied of the neurotrophins. Numerous studies have tested in vivo the effects of either administering exoge-nous NGF or sequestering naturally produced NGF. These experiments show that it can alter neuronal gene expres-sion in sensory neurons (e.g., Michael et al 1997; Woolf 1996). Inflammatory injuries cause increased levels of NGF in peripheral tissues and therefore increase the exposure of sensory neurons to NGF (McMahon et al 1995; Woolf et al 1994; Woolf 1996). Under these conditions, NGF-dependent changes occur in the
excitabil-ity and in the responsiveness of neurons to stimuli. Underlying these effects are alterations in neurotransmitter and neuropeptide release and in the sensitivity of neurons by alterations in neurotransmitter receptors and ion chan-nels (e.g.; Fjell et al 1999; Malcangio et al 1997; McMa-hon 1996; Woolf et al 1996). Furthermore, NGF can induce morphologic changes, such as sprouting and out-growth in responsive sensory or sympathetic axons, par-ticularly after injury (e.g., Ramer et al 2000). These effects are all consistent with increased retrograde transport of trkA signaling complexes that alter gene expression in the NGF-responsive neurons.
One of the unexpected discoveries from these experi-ments was that NGF could produce changes in neuronal excitability and behaviors that are inconsistent with the time course of receptor internalization, retrograde trans-port, and gene expression. One example of this phenom-enon comes from a model of chemical inflammation of the rat bladder. About 30 min after exposure of the bladder to an irritant, there is a behavioral and electrophysiologic response corresponding to pain. This rapid response, analogous to pain in humans, is dependent on NGF, confined to the population of sensory nerve fibers that express trkA, and involves the rapid induction of NGF mRNA and protein in the inflamed tissue (Dmitrieva et al 1997; Oddiah et al 1998). These results strongly suggest that NGF has direct and rapid actions on neuronal excit-ability and that these effects occur too quickly to be mediated by retrograde transport of signaling complexes.
NGF and the Basal Forebrain
Cholinergic System
In contrast to the periphery, NGF expression in the central nervous system is much more restricted; NGF mRNA and protein are expressed in a number of brain regions, with the hippocampus providing the single largest source of NGF in the entire CNS (Korsching et al 1985). In the hippocampus, NGF messenger RNA and protein is ex-pressed by the principal excitatory (glutamate) neurons in the hippocampus, as well as by a subset ofg-aminobutyric acid– containing inhibitory neurons (Rocamora et al 1996). These hippocampal target cells receive rich inner-vation from ascending neurons with their cell bodies in the basal forebrain.
noncho-linergic neurons that express trkA, including some of the principal cells of the hippocampus (Cellerino 1996; Holtz-man et al 1995). There are no noncholinergic neurons known to express p75NTR, but this receptor is expressed by some hippocampal astrocytes (Dougherty and Milner 1999). The distribution of NGF and its receptors suggests an intimate and precise interrelationship between cholin-ergic neurons and their NGF-producing targets.
Given that NGF is expressed in the targets for cholinergic neurons and that cholinergic neurons are responsive to NGF, it may be expected that NGF would have a classic neurotro-phic role to mediate cholinergic cell survival during devel-opment; however, studies using transgenic mice deficient in NGF or trkA suggest that in prenatal and early postnatal periods there is little, if any, cholinergic cell loss in the CNS (Chen et al 1997; Crowley et al 1994; Fagan et al 1997; Ruberti et al 2000). This absence of central cholinergic deficits contrasts with the notable absence of sensory neurons observed in the NGF knockout transgenic animals prenatally and postnatally (Crowley et al 1994). These results clearly show that NGF is not critical for basal forebrain cholinergic neuron survival during development. Although basal fore-brain cholinergic neurons are responsive to NGF during their development, other trophic factors are required for their survival. The actual survival factors are unknown, but it is clear that developing cholinergic neurons are responsive to a number of trophic factors, particularly BDNF (Ward and Hagg 2000) and maintain this responsiveness in adult life (Koliatsos et al 1994).
To what extent NGF is necessary as a survival factor for adult cholinergic neurons remains controversial. It pro-motes the survival of cholinergic neurons after axotomy (Hefti 1986; Kromer 1987; Williams et al 1986). Measur-ing the effects of reduced availability of NGF to undam-aged cholinergic neurons has been technically difficult. Partial removal in vivo of the NGF supply to cholinergic neurons in transgenic animals or near-complete removal by ablation of the hippocampus causes severe cholinergic neuronal atrophy, with little or no neuronal death (Chen et al 1997; Sofroniew et al 1993). These studies have been interpreted to mean that NGF is not a survival factor for cholinergic neurons in the adult; however, a more recent study using a transgenic mouse that lacks NGF and survives to adulthood suggests that a complete lack of NGF in vivo in the adult does cause cholinergic cell loss in the basal forebrain (Ruberti et al 2000).
As outlined above, reduction in the NGF supply to basal forebrain cholinergic neurons causes atrophy in cholin-ergic neurons. These changes are reminiscent of the cholinergic neuronal changes in Alzheimer’s disease (AD). In AD, however, there is no evidence that it is NGF deficiency that causes cholinergic cell loss. In fact, mea-surements in postmortem brains from AD patients show
that there is no change in the capacity of the CNS to produce NGF (mRNA levels) with increased levels of NGF protein in the cortex and hippocampus (Fahnestock et al 1996; Hock et al 1998; Jette et al 1994; Scott et al 1995). Increased NGF levels are thought to reflect a decreased ability of cholinergic neurons to retrogradely transport NGF.
In the adult, NGF influences the messenger RNA levels, structural plasticity, response to injury, and maintenance of basal forebrain cholinergic neurons. Administration of NGF to the uninjured CNS causes a number of effects on cholin-ergic neurons, including hypertrophy, sprouting, upregulation of NGF receptors, increased levels of choline acetyltrans-ferase (ChAT), and increased choline uptake (Heisenberg et al 1994; Higgins et al 1989; Lapchak et al 1992; Mobley et al 1985). These results are all consistent with a role of NGF to maintain the cholinergic phenotype through retrograde transport of the NGF/trkA signaling complex from cholin-ergic nerve terminals in the hippocampus to the cell bodies in the basal forebrain (Seiler et al 1984).
There are also rapid actions of NGF on cholinergic neurons. It can rapidly increase acetylcholine release and choline uptake in synaptosomes (Knipper et al 1994), an effect that cannot include effects via gene expression because the cell bodies are not present in these preparations. Simi-larly, in cultured septal neurons, NGF has been shown to produce rapid increases in intracellular calcium levels, a stimulus known to enhance neurotransmitter release (Nonner et al 2000). It has been shown to cause rapid increases in the firing rate of cholinergic neurons (Albeck et al 1999). In other types of neurons, NGF can cause rapid activation of calcium-dependent potassium channels (Holm et al 1997) and volt-age-sensitive calcium channels (Jia et al 1999), actions that may underlie NGF effects on cholinergic neuronal excitabil-ity and acetylcholine release. These findings fit well with the emerging evidence that neurotrophins can behave as neuro-transmitter-like molecules. In fact, in a similar manner to a neurotransmitter, NGF can be released by neurons upon depolarization (Blo¨chl and Thoenen 1995).
Nerve Growth Factor, Cognition, and
Alzheimer’s Disease
On the basis of the data described above, NGF, through its effects on the cholinergic system, may be predicted to improve cognitive functions in AD. Animal studies strongly support this notion and have shown that NGF can enhance performance in behavioral cognitive tasks (Gustilo et al 1999; Pellymounter et al 1996) and slow cognitive decline in aged rats (Martinez-Serrano and Bjorklund 1998). It has been shown to have a role in physiologic correlates of memory acquisition in the hip-pocampus (Bergado et al 1998). Conversely, reduction of NGF levels appears to have a detrimental effect on learning and cognitive behaviors. Animals with reduced levels of NGF (anti-NGF antibodies, transgenic animals) show impaired spatial learning (Chen et al 1997; Van der Zee et al 1995).
From the evidence outlined above, it is likely that increased exposure of NGF to cholinergic neurons will be beneficial in AD. Because NGF does not cross the blood– brain barrier, and there are no small molecule agonists available, at present NGF must be delivered directly to the central nervous system. Experimental meth-ods include gene transfer, cell grafts, or direct administra-tion of NGF through intracerebroventricular infusion (Ea-gle et al 1995; Gustilo et al 1999; Hefti 1986; Kromer 1987; Martinez-Serrano and Bjorklund 1998; Williams et al 1986; Winkler et al 2000; Wyman et al 1999). In general, in animals with deafferentation lesions and im-paired cognitive function, these approaches have been shown to enhance cholinergic neuron integrity and restore some behavioral functions.
In AD patients, there is limited evidence at present that NGF may be beneficial. Treatment with NGF was tested in three patients, with no clear improvement in cognitive function (Eriksdotter Jonhagen et al 1998). In this study, the potential therapeutic benefits of NGF infusion were offset by severe pain. Large quantities of NGF adminis-tered over long periods are likely to diffuse to affect the peripheral nervous system and cause pain. These periph-eral side effects at present limit the therapeutic use of intracerebroventricular NGF. In addition to the painful side effects in humans, recent animal studies document a number of adverse behavioral effects and Schwann cell proliferation after high doses of NGF administered by the intracerebroventricular route (Winkler et al 2000). On this basis, alternative means of delivering NGF to the CNS must be considered. In the future, gene therapy and transplantation techniques may prove successful, but these strategies are confined to the laboratory at present.
It may be possible to use pharmacologic approaches to increase the production of NGF within the CNS. The
potential advantages of this strategy are that NGF should be induced in the correct brain regions. In addition, this approach is likely to circumvent the peripheral side effects of NGF and would not require surgical procedures. Nerve growth factor mRNA expression and protein secretion are tightly linked and can be regulated by a variety of compounds in vitro, including known drugs that act on neurotransmitter receptors (reviewed in Bennett et al 1999). Therefore, numerous agents have the potential to enhance NGF expression and secretion in the brain.
A number of nonclassic compounds have shown to increase synthesis of NGF in astrocytes. These drugs include a phosphodiesterase inhibitor, propentofylline (Rother et al 1998) and various quinone derivatives, such as idebenone, pyrroloquinoline quinone, and 1,4-benzo-quinone (Takeuchi et al 1990; Yamada et al 1999; Yamaguchi et al 1993). It is argued that these drugs may improve cholinergic neuron survival and enhance cogni-tive performance via increasing astrocyte secretion of NGF. How these drugs modulate NGF levels is unknown, and it is not known whether their clinical or preclinical efficacy in AD (if any) resides specifically in the ability of these compounds to increase NGF levels.
Cholinergic and Glutamatergic Regulation
of NGF Levels: Prospects for AD Treatment
In AD there is extensive loss of cholinergic and glutama-tergic input into the hippocampus (reviewed in Francis et al 1993, 1999). Loss of nicotinic acetylcholine receptors is a clear finding in AD (Court 2001; Nordberg 2001), suggesting that fast cholinergic transmission is particularly affected in the disease. In the hippocampus, nicotinic receptors effect multiple neurotransmitter systems, which may underlie the cognitive enhancing effects of nicotinic receptor agonists and opposite effects of nicotinic antag-onists. In addition to effects on classical neurotransmitter systems, it is possible that the cholinergic and glutamater-gic inputs may be endogenous regulators of NGF in the hippocampus—and therefore that hippocampal NGF can be regulated by conventional and novel drugs targeted to the cholinergic or glutamatergic systems.
transection) is used, which cuts the axons of both the cholinergic and noncholinergic septohippocampal neu-rons, there is a long-lasting downregulation of NGF mRNA in the hippocampus (Table 1). Surgery to reduce glutamate input into the hippocampus produces upregula-tion of NGF levels (Table 1). These studies broadly suggest that CNS lesions mimicking the hippocampal deafferentation in AD lead to upregulation of NGF levels in targets and that NGF levels are subject to complex control mechanisms. It is difficult to extrapolate these findings into AD, where there is little if any change in NGF mRNA, but rather enhanced levels of protein due to impaired retrograde transport of NGF.
Because the lesion studies suggest that cholinergic and glutamatergic afferents regulate NGF production in the hippocampus, cholinergic drugs, glutamatergic drugs, or both may be effective inducers of NGF. Nicotinic drugs, as well as drugs that enhance acetylcholine transmission, are effective in improving cognitive function in both normal humans and in AD patients (Francis et al 1999; Newhouse 2001), and there is the interesting possibility that they may influence NGF. Perhaps surprisingly, there are few studies to have looked at this possibility in vivo. A recent study has tested the effects of local administration of cholinergic drugs into the hippocampus on neurotrophin mRNA levels (French et al 1999). Drug doses were carefully chosen in relation to previous studies, and only doses that caused no hippocampal neuronal death and did not cause seizures were used.
Nicotine, when injected into the hippocampus, caused a persistent increase in NGF mRNA levels in the principal neurons of the hippocampus that emerged 2 to 4 hours after drug administration and was still apparent 72 hours after drug administration, the last time point analyzed in the study. This contrasted with the muscarinic receptor
agonist pilocarpine, which produced a very transient increase in NGF mRNA levels (French et al 1999). The effect was also highly selective for NGF, because BDNF mRNA levels showed a rapid and transient increase after nicotine administration (French et al 1999). The nicotine-induced increase of NGF mRNA levels was blocked by N-methyl-D-aspartate and a -amino-3-hydroxy-5-methyl-4-isoxazole propionate glutamate receptor antagonists, consistent with the increase being dependent on nicotine-induced release of glutamate within the hippocampus (Gray et al 1996). The study strongly suggests that quite large and long-lasting induction of NGF in appropriate
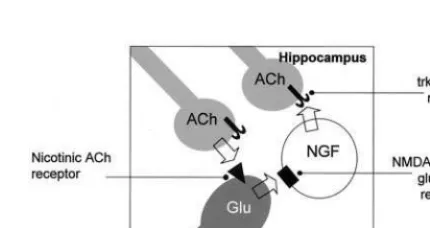
Figure 2. Nicotinic–nerve growth factor (NGF) interactions in the hippocampus. Current evidence suggests that local infusions of nicotine into the hippocampus increases NGF messenger RNA levels. The results are consistent with the model whereby acetylcholine (ACh) is released from nerve terminals to activate nicotinic receptors on glutamatergic neurons. Glutamate is re-leased and viaa-amino-3-hydroxy-5-methyl-4-isoxazole
propi-onate (AMPA), N-methyl-D-aspartate (NMDA) receptors, or both, it stimulates NGF production in cells intrinsic to the hippocampus. Nerve growth factor will activate trkA receptors on the cholinergic nerve terminals, providing trophic support and influencing the activity of these neurons to further enhance ACh release.
Table 1. Afferent Regulation of Nerve Growth Factor (NGF) Levels in the Hippocampus
Deafferentation lesion Effect Reference
AF64A lesion of
septohippocampal cholinergic neurons
1NGF protein and mRNA 7 weeks after lesion
Hellweg et al 1997
Ig92 saporin lesion of septohippocampal cholinergic neurons
1NGF protein, 3 months after lesion
1NGF protein, 2 weeks after lesion No change NGF mRNA
1 week–5 months after lesion Blocks seizure-induced increases in NGF
Gu et al 1998 Roßner et al 1997 Yu et al 1995
Kokaia et al 1996
Fimbria fornix transection removing cholinergic and other inputs
2NGF mRNA da Penha Berzaghi et al 1993 Lapchak et al 1992 Gasser et al 1986
1NGF protein 10 d after lesion Larkfors et al 1987 Angular bundle transection
and/or entorhinal cortex lesion
1NGF mRNA
1NGF mRNA (rapid)
1NGF protein 3,8 d after lesion
CNS regions can be achieved with drug doses that are pharmacologically relevant.
These findings suggest a model of reciprocal interaction between cholinergic afferents and NGF-producing princi-pal neurons in the hippocampus (Figure 2), an idea developed by other researchers (e.g., Knipper et al 1994; Yu et al 1995), which provides a testable concept for future studies. The model shown in Figure 2 defines a reinforcing loop whereby acetylcholine release elevates NGF production, and then NGF affects acetylcholine neurons to provide trophic support and further enhance acetylcholine synthesis and release. The implications of this model are that some cholinergic drugs may have cognitive enhancing effects beyond their primary effects as cholinesterase inhibitors and receptor agonists. A num-ber of cholinergic drugs, including nicotinic receptor agonists, are currently in development for use in AD (Francis et al 1999). It is possible that in addition to having direct effects on cognition (e.g., Newhouse 2001), this kind of drug may provide a positive neurotrophic influ-ence to the cholinergic neurons that may help both to prevent their degeneration in AD and to enhance cholin-ergic neurotransmission.
Some of the work presented in this article was supported by the Wellcome Trust and the Medical Research Council of Great Britain.
I gratefully acknowledge my collaborators on these projects: Michael V. Sofroniew, John V. Priestley, and Stephen B. McMahon.
Aspects of this work were presented at the symposium “Nicotine Mechanisms in Alzheimer’s Disease,” March 16 –18, 2000, Fajardo, Puerto Rico. The conference was sponsored by the Society of Biological Psychiatry through an unrestricted educational grant provided by Janssen Pharmaeutica LP.
References
Albeck DS, Backman C, Veng L, Friden P, Rose GM, Granholm A (1999): Acute application of NGF increases the firing rate of aged rat basal forebrain neurons.Eur J Neurosci11:2291– 2304.
Batchelor PE, Armstrong DM, Blaker SN, Gage FH (1989): Nerve growth factor receptor and choline acetyltransferase colocalization in neurons within the rat forebrain: Response to fimbria-fornix transection.J Comp Neurol284:187–204. Bennett DLH, McMahon SB, Rattray M, Shelton DL (1999):
Nerve growth factor and sensory nerve function. In: Brain SD, Moore PK, editors.Pain and Neurogenic Inflammation.
Basel, Switzerland: Birka¨user Verlag, 167–193.
Bergado JA, Gomez-Soria AA, Cruz R, Fernandez CI (1998): Nerve growth factor improves evoked potentials and long-term potentiation in the dentate gyrus of presenile rats.Eur J Pharmacol345:181–184.
Blochl A, Thoenen H (1995): Characterization of nerve growth factor (NGF) release from hippocampal neurons: Evidence for a constitutive and an unconventional sodium-dependent regulated pathway.Eur J Neurosci7:1220 –1228.
Cellerino A (1996): Expression of messenger RNA coding for the nerve growth factor receptor trkA in the hippocampus of the adult rat.Neuroscience70:613– 616.
Chao MV (1994): The p75 neurotrophin receptor.J Neurobiol
25:1373–1385.
Chen KS, Nishimura MC, Armanini MP, Crowley C, Spencer SD, Phillips HS (1997): Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J Neurosci 17: 7288 –7296.
Conner JM, Fass-Holmes B, Varon S (1994): Changes in nerve growth factor immunoreactivity following entorhinal cortex lesions: Possible molecular mechanism regulating cholinergic sprouting.J Comp Neurol345:409 – 418.
Court J, Martin-Ruiz C, Piggott M, Spurden D, Griffiths M, Perry E (2001): Nicotinic receptor abnormalities in Alzhei-mer’s disease.Biol Psychiatry49:175–184.
Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, et al (1994): Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons.Cell76:1001– 1011.
da Penha Berzaghi M, Cooper J, Castren E, Zafra F, Sofroniew M, Thoenen H, et al (1993): Cholinergic regulation of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) but not neurotrophin-3 (NT-3) mRNA levels in the developing rat hippocampus.J Neurosci13:3818 –3826. Dmitrieva N, Shelton D, Rice ASC, McMahon SB (1997): The
role of nerve growth factor in a model of visceral inflamma-tion.Neuroscience78:449 – 459.
Dougherty KD, Milner TA (1999): P75NTR immunoreactivity in the rat dentate gyrus is mostly within presynaptic profiles but is also found in some astrocytic and postsynaptic profiles.
J Comp Neurol407:77–91.
Eagle KS, Chalmers GR, Clary DO, Gage FH (1995): Axonal regeneration and limited functional recovery following hip-pocampal deafferentation.J Comp Neurol363:377–388. Eriksdotter Jonhagen M, Nordberg A, Amberla K, Backman L,
Ebendal T, Mayerson B, et al (1998): Intracerebral infusion of nerve growth factor in three patients with Alzheimer’s dis-ease.Dement Geriatr Cogn Disord9:246 –257.
Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM (1997): A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J Neurosci
17:7644 –7654.
Fahnestock M, Scott SA, Jette N, Weingartner JA, Crutcher KA (1996): Nerve growth factor mRNA and protein levels mea-sured in the same tissue from normal and Alzheimer’s disease parietal cortex.Mol Brain Res42:175–178.
Fjell J, Cummins TR, Fried K, Black JA, Waxman SG (1999): In vivo NGF deprivation reduces SNS expression and TTX-R sodium currents in IB4-negative DRG neurons. J Neuro-physiol81:803– 810.
Francis PT, Sims NR, Procter AW, Bowen DM (1993): Cortical pyramidal neurone loss may cause glutamatergic hypoactivity and cognitive impairment in Alzheimer’s disease: Investiga-tive and therapeutic perspecInvestiga-tives. J Neurochem 60:1589 – 1604.
Freidman WJ, Greene LA (1999): Neurotrophin signaling via Trks and p75.Exp Cell Res253:131–142.
French SJ, Humby T, Horner CH, Sofroniew MV, Rattray M (1999): Hippocampal neurotrophin and trk receptor mRNA levels are altered by local administration of nicotine, carba-chol and pilocarpine.Brain Res Mol Brain Res67:124 –136. Gasser UE, Weskamp G, Otten U, Dravid AR (1986): Time course of the elevation of nerve growth factor (NGF) content in the hippocampus and septum following lesions of the septohippocampal pathway in rats.Brain Res376:351–356. Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA (1996):
Hippocampal synaptic transmission enhanced by low concen-trations of nicotine.Nature383:713–716.
Grimes ML, Zhou J, Beattle EC, Yuen EC, Hall DE, Valletta JS, et al (1996): Endocytosis of activated TrkA: Evidence that nerve growth factor induces formation of signaling endo-somes.J Neurosci16:7950 –7964.
Gu Z, Yu J, Perez-Polo R (1998): Long term changes in brain cholinergic markers and nerve growth factor levels after partial immunolesion.Brain Res801:190 –197.
Gustilo MC, Markowska AL, Breckler SJ, Fleischman CA, Price DL, Koliatsos VE (1999): Evidence that nerve growth factor influences recent memory through structural changes in septohippocampal cholinergic neurons.J Comp Neurol405: 491–507.
Gwag BJ, Sessler F, Kimmerer K, Springer JE (1994): Neuro-trophic factor mRNA expression in the dentate gyrus is increased after angular bundle transection.Brain Res647:23– 29.
Hefti F (1986): Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transactions. J Neurosci
6:2155–2162.
Heisenberg CP, Cooper JD, Berke J, Sofroniew MV (1994): NMDA potentiates NGF-induced sprouting of septal cholin-ergic fibres.Neuroreport12:413– 416.
Hellweg R, Humpel C, Lowe A, Hortnagl H (1997): Moderate lesion of the rat cholinergic septohippocampal pathway in-creases hippocampal nerve growth factor synthesis: Evidence for long-term compensatory changes?Mol Brain Res45:171– 181.
Higgins GA, Koh S, Chen KS, Gage FH (1989): NGF induction of NGF receptor gene expression and cholinergic neuron hypertrophy within the basal forebrain of the adult rat.
Neuron3:247–256.
Hock C, Heese K, Muller-Spahn F, Hulette C, Rosenberg C, Otten U (1998): Decreased trkA neurotrophin receptor ex-pression in the parietal cortex of patients with Alzheimer’s disease.Neurosci Lett241:151–154.
Holm NR, Christophersen P, Olesen SP, Gammeltoft S (1997): Activation of calcium-dependent potassium channels in mouse brain neurons by neurotrophin-3 and nerve growth factor.Proc Natl Acad Sci U S A94:1002–1006.
Holtzman DM, Kilbridge J, Li Y, Cunningham ET Jr, Lenn NJ, Clary DO, et al (1995): TrkA expression in the CNS:
Evidence for the existence of several novel NGF-responsive CNS neurons.J Neurosci15:1567–1576.
Jette N, Cole MS, Fahnestock M (1994): NGF mRNA is not decreased in frontal cortex from Alzheimer’s disease patients.
Mol Brain Res25:242–250.
Jia M, Li M, Liu XW, Jiang H, Nelson PG, Guroff G (1999): Voltage-sensitive calcium currents are acutely increased by nerve growth factor in PC12 cells.J Neurophysiol82:2847– 2852.
Knipper M, da Penha Berzaghi M, Blochl A, Breer H, Thoenen H, Lindholm D (1994): Positive feedback between acetylcho-line and the neurotrophins nerve growth factor and brain derived neurotrophic factor in the hippocampus.Eur J Neu-rosci4:668 – 671.
Kokaia M, Ferencz I, Leanza G, Elmer E, Metsis M, Kokaia Z, et al (1996): Immunolesioning of basal forebrain cholinergic neurons facilitates hippocampal kindling and perturbs neuro-trophin messenger RNA regulation. Neuroscience 70:313– 327.
Koliatsos VE, Price DL, Gouras GK, Cayouteete MH, Burton LE, Winslow JW (1994): Highly selective effects of nerve growth factor, brain-derived neurotrophic factor, and neuro-trophin-3 on intact and injured basal forebrain magnocellular neurons.J Comp Neurol343:247–262.
Korsching S, Auburger G, Heumann R, Scott J, Thoenen H (1985): Levels of nerve growth factor and its mRNA in the central nervous system of the rat correlate with cholinergic innervation.EMBO J4:1389 –1393.
Kromer LF (1987): Nerve growth factor treatment after brain injury prevents neuronal death.Science235:214 –216. Lapchak PA, Jenden DJ, Hefti F (1992): Pharmacological
stim-ulation reveals recombinant human nerve growth factor-induced increases of in vivo hippocampal cholinergic func-tion measured in rats with partial fimbria transacfunc-tions.
Neuroscience50:847– 856.
Larkfors L, Stromberg I, Ebendal T, Olson L (1987): Nerve growth factor protein level increases in the adult rat hip-pocampus after a specific cholinergic lesion.J Neurosci Res
18: 525–531.
Lindsay RM, Widgand SJ, Altar CA, DiStephano PS (1994): Neurotrophic factors: From molecule to man.Trends Neuro-sci17:182–190.
Malcangio M, Garrett NE, Cruwys S, Tomlinson DR (1997): Nerve growth factor- and neurotrophin-3-induced changes in nociceptive threshold and the release of substance P from the rat isolated spinal cord.J Neurosci17:8459 – 8467.
Martinez-Serrano A, Bjorklund A (1998): Ex vivo nerve growth factor gene transfer to the basal forebrain in presymptomatic middle-aged rats prevents the development of cholinergic neuron atrophy and cognitive impairment during aging.Proc Natl Acad Sci U S A95:1858 –1863.
McDonald NQ, Blundell TL (1991): Crystallization and charac-terization of the high molecular weight form of nerve growth factor (7 S NGF).J Mol Biol219:595– 601.
McMahon SB (1996): NGF as a mediator of inflammatory pain.
Philos Trans R Soc Lond B Biol Sci351:431– 440.
Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV (1997): Nerve growth factor treatment in-creases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci 17:8476 – 8490.
Mobley W, Rutkowski J, Tennekoon G, Buchanan K, Johnston M (1985): Choline acetyltransferase activity in striatum of neonatal rats increased by nerve growth factor. Science
229:284 –287.
Newhouse PA, Potter A, Kelton M, Corwin J (2001): Nicotinic treatment of Alzheimer’s disease. Biol Psychiatry 49:268 – 278.
Nonner D, Barrett EF, Barrett JN (2000): Brief exposure to neurotrophins produces a calcium-dependent increase in cho-line acetyltransferase activity in cultured rat septal neurons.
J Neurochem74:988 –999.
Nordberg A (2001): Nicotinic receptor abnormalities of Alzhei-mer’s disease: Therapeutic implications.Biol Psychiatry49: 200 –210.
Oddiah D, Anand P, McMahon SB, Rattray M (1998): Rapid increase of NGF, BDNF and NT-3 mRNAs in inflamed bladder.Neuroreport9:1455–1458.
Pellymounter MA, Cullen MJ, Baker MB, Gollub M, Wellman C (1996): The effects of intrahippocampal NGF on spatial learning in aged Long-Evans rats. Mol Chem Neuropathol
29:211–226.
Ramer MS, Priestley JV, McMahon SB (2000): Functional regeneration of sensory axons into the adult spinal cord.
Nature403:312–316.
Riccio A, Pierchala BA, Ciarallo CL, Ginty DD (1997): An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons.Science277:1097–1100. Rocamora N, Pascual M, Acsady L, de Lecea L, Freund TF,
Soriano E (1996): Expression of NGF and NT3 mRNAs in hippocampal interneurons innervated by the GABA septohip-pocampal pathway.J Neurosci16:3991– 4004.
Roßner S, Wortwein G, Gu Z, Yu J, Schleibs R, Bigl V, et al (1997): Cholinergic control of nerve growth factor in adult rats: Evidence from cortical cholinergic deafferentation and chronic drug treatment.J Neurochem69:947–953.
Rother M, Erkinjuntti T, Roessner M, Kittner B, Marcusson J, Karlsson I (1998): Propentofylline in the treatment of Alz-heimer’s disease and vascular dementia: A review of phase III trials.Dement Geriatr Cogn Disord9(suppl 1):36 – 43. Ruberti F, Capsoni S, Comparini A, Di Daniel E, Franzot J,
Gonfloni S, et al (2000): Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy.J Neurosci20:2589 – 2601.
Scott SA, Mufson EJ, Weingartner JA, Skau KA, Crutcher KA (1995): Nerve growth factor in Alzheimer’s disease: In-creased levels throughout the brain coupled with declines in nucleus basalis.J Neurosci15:6213– 6221.
Seiler M, Schwab ME (1984): Specific retrograde transport of nerve growth factor from neocortex to nucleus basalis in the rat.Brain Res300:33–39.
Snider WD, McMahon SB (1998): Tackling pain at the source: New ideas about nociceptors.Neuron20:629 – 632. Sofroniew MV, Cooper JD, Svendsen CN, Crossman P, Ip NY,
Lindsay RM, et al (1993): Atrophy but not death of adult septal cholinergic neurons after ablation of target capacity to produce mRNAs for NGF, BDNF, and NT3. J Neurosci
13:5263–5276.
Takeuchi R, Murase K, Furkawa Y, Furukawa S, Hayashi K (1990): Stimulation of nerve growth factor synthesis/secre-tion by 1,4-benzoquinone and its derivatives in cultured mouse astroglial cells.FEBS Lett261:63– 66.
Ultrech MH, Wiesmann C, Simmons LC, Henrich J, Yang M, Reilly D, et al (1999): Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. J Mol Biol
290:149 –159.
Van der Zee CE, Lourenssen S, Stanisz J, Diamond J (1995): NGF deprivation of adult rat brain results in cholinergic hypofunction and selective impairments in spatial learning.
Eur J Neurosci7:160 –168.
Ward NL, Hagg T (2000): BDNF is needed for postnatal maturation of basal forebrain and neostriatum cholinergic neurons in vivo.Exp Neurol162:297–310.
Williams LR, Varon S, Peterson GM, Wictorin K, Fischer W, Bjorklund A, Gage FH (1986): Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc Natl Acad Sci U S A
83:9231–9235.
Winkler J, Ramirex GA, Thal LJ, Waite JJ (2000): Nerve growth factor (NGF) augments cortical and hippocampal cholinergic functioning after p75NGF receptor-mediated deafferentation but impairs inhibitory avoidance and induces fear-related behaviours.J Neurosci20:834 – 844.
Woolf CJ (1996): Phenotypic modification of primary sensory neurons: The role of nerve growth factor in the production of persistent pain.Philos Trans R Soc Lond B Biol Sci35:441– 448.
Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J (1994): Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience
62:327–331.
Wyman T, Roherer D, Kirigiti P, Nichols H, Pilcher K, Nilaver G, et al (1999): Promoter-activated expression of nerve growth factor for treatment of neurodegenerative diseases.
Gene Ther6:1648 –1660.
Yamada K, Tanaka T, Han D, Senzaki K, Kameyama T, Nabeshima T (1999): Protective effects of idebenone and alpha-tocopherol on beta-amyloid-(1-42)-induced learning and memory deficits in rats: Implication of oxidative stress in beta-amyloid-induced neurotoxicity in vivo. Eur J Neurosci
11:83–90.
Yamaguchi K, Sasano A, Urakami T, Tsuji T, Kondo K (1993): Stimulation of nerve growth factor production by pyrrolo-quinoline quinone and its derivatives in vitro and in vivo.
Biosci Biotechnol Biochem57:1231–1233.