Inhibitors of HMG-CoA reductase sensitize human smooth muscle
cells to Fas-ligand and cytokine-induced cell death
Anita C. Knapp
a, Jinwen Huang
a, Gary Starling
b, Peter A. Kiener
b,*
aDepartment of Metabolic Research,Bristol-Myers Squibb,PRI,PO Box 4000,Princeton,NJ 08543, USA bDepartment of Immunology,Bristol-Myers Squibb,PRI,K14-09,PO Box 4000,Princeton,NJ 08543, USA
Received 30 June 1999; received in revised form 23 September 1999; accepted 3 November 1999
Abstract
Hydroxymethylglutaryl CoA (HMG CoA) reductase inhibitors, or statins, have been shown to reduce atherosclerotic cardiovascular morbidity and mortality. Atherosclerotic plaque lesions can be chronically inflamed and vulnerable to rupture or stable and less rupture-prone. Human smooth muscle cells (SMC) are critically important in maintaining the stability of atherosclerotic plaques. This stability may be greatly influenced by pro-inflammatory mediators such as IFN-g, TNF-a, and Il-1b
and Fas ligand (FasL) that are present in human atheroma. The purpose of the present study was to examine the effect of the statins on apoptosis of SMC. We have found that SMC are normally resistant to Fas or cytokine-induced apoptosis, but can be sensitized to these agents with pharmacological concentrations of some statins. Simvastatin and lovastatin strongly sensitized the cells to apoptotic agents while atorvastatin was less effective. In contrast to the lipophilic statins, the hydrophilic statin pravastatin did not induce this sensitization of SMC to apoptosis. Treatment of SMC with either mevalonate, the product of the HMG-CoA reductase, or geranylgeranylpyrophosphate, a down stream intermediate, prevented lipophilic statin-induced sensitization to apoptosis. These results suggest that prenylation of one or more proteins is critically involved in regulating the sensitivity of SMC to apoptotic stimuli. Our data support the emerging evidence that through this pathway the various statins may have effects which are beyond a simple lowering of the levels of circulating cholesterol. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Hydroxymethylglutaryl CoA (HMG CoA) reductase inhibitors; Cell death; Smooth muscle cells; FasL; Cytokines
www.elsevier.com/locate/atherosclerosis
1. Introduction
A high level of circulating cholesterol in patients is one of the risk factors for the development of coronary atherosclerotic lesions. To lower plasma lipid levels the rate limiting step in cholesterol biosynthesis is inhibited with hydroxymethylglutaryl CoA (HMG CoA) reduc-tase inhibitors (statins) [1 – 3]. In contrast to their lipid lowering mechanism, it is less clear how the different drugs may directly affect the atherosclerotic lesion and what actually triggers rupture of the atherosclerotic plaque [4]. Cardiac infarction occurs when a final thrombosis occludes the blood vessel, but this is
pre-ceded by many benign thrombotic events that probably occur throughout the lifetime of the lesion [5]. Thus, it is important to understand how the stability of the advanced, vulnerable plaque is influenced by the vari-ous cell types and soluble mediators, such as the statins. Recent studies, both in vitro and in vivo, have demonstrated that some statins are able to induce changes in cells or tissues that are unrelated to reduc-tion of plasma cholesterol [6 – 12]. Since these effects were reversed with mevalonate and other metabolites of the isoprene synthesis pathway, protein prenylation seems to play an important role [13]. Furthermore, chronic inflammation significantly contributes to the development of atherosclerotic plaques [4,14 – 16]. As shown by histochemical analysis, highly vulnerable and ruptured human plaques contain more macrophages and fewer smooth muscle cells (SMC) [5]. Additionally, an enhanced expression of inflammatory and pro-* Corresponding author. Tel.:+1-609-252-5151; fax:+
1-609-252-6058.
E-mail address:[email protected] (P.A. Kiener).
apoptotic mediators correlates with plaque instability [17,18]. These pro-inflammatory mediators are able to induce apoptosis of SMC in vitro [19], and SMC in the advanced plaque may undergo increased programmed cell death. One pathway that appears to be involved in apoptosis is the Fas (CD95 or Apo-1)-FasL system. Fas is a type I membrane protein belonging to the TNF receptor family and its receptor mediates a death signal after binding to its ligand, FasL. Fas expression has been found in the human atherosclerotic intima [20], indeed, most of the cells in the advanced lesion have been found to express Fas [21]. In addition, activated T-cells and monocytes are major sources for both FasL and cytokines, and these cells are present at sites of plaque rupture in human atheroma [14]. Overall, the findings from many investigators indicate that the dy-namics of an atherosclerotic plaque are influenced by the different proinflammatory mediators, such as IFN-g, TNF-a, Il-1band FasL, that are present in the local microenvironment [14].
In the advanced, highly inflamed plaque, many dif-ferent cell types, including SMC and macrophages, can undergo apoptosis. However, there is only a rudimen-tary understanding how plaque stability is affected through apoptosis of the various cells. Although in the human atherosclerotic plaque, both necrosis and apop-tosis have been localized to sites of plaque rupture [22], abundant evidence suggests that SMC primarily die by apoptosis [23]. For example, in contrast to SMC from normal vessels, cells derived from human atheroscle-rotic plaques proliferate more slowly, undergo fewer proliferative cycles, and are more susceptible to sponta-neous apoptotic cell death [23,24]. In addition, it has been shown that intimal lesions contain more TUNEL positive SMC than the non-atherosclerotic arterial in-tima [25]. Because the production of plaque stabilizing extracellular matrix proteins in the advanced lesion is considered to be beneficial and is primarily dependent on SMC in the intima, depletion of SMC in the ad-vanced plaque is thought to enhance plaque thinning [5,21,23 – 28]. However, SMC may play different roles in different pathological contexts. Overall, the pro-infl-ammatory environment of a plaque may increase the susceptibility of SMC to cell death, and the concomi-tant loss of synthesis of extracellular matrix proteins would then lead to an increase in plaque instability [5,29].
The effects of the different statins in the pro-inflam-matory or pro-apoptotic environment of the plaque is not well understood, but could have important implica-tions in cardiovascular disease. In this report we demonstrate that lipophilic statins at in vitro concentra-tions that are potentially pharmacologically relevant, directly sensitize SMC to both FasL and cytokine in-duced apoptosis.
2. Experimental procedures
2.1. Reagents
Simvastatin, lovastatin and pravastatin were synthe-sized by Bristol-Myers Squibb and atorvastatin was a generous gift from Parke-Davis. Simvastatin, lovastatin and mevalonate were activated to their active open-ring forms as described [30]. Geranylgeranylpyrophosphate and farnesylpyrophosphate were obtained from Alexis Biochemicals and were used at 20mmol/l. Recombinant soluble human Fas-ligand (sFasL) and human Fas-Ig fusion proteins were produced and purified as described previously [31]. The sFasL fusion protein contains the extracellular domain of human FasL and the extracellu-lar region of mouse CD8. Analysis of the purified fusion protein by gel filtration revealed that it is a mixture of dimers and trimers. As such this protein very effectively stimulates apoptosis in Jurkat cells and addi-tional crosslinking of the fusion protein with an anti-mouse CD8 mAb did not further enhance activity. Recombinant human TNF-a, IFN-g, and Il-1b were obtained from R&D Systems. sFasL was used at 100 ng/ml and Fas-Ig at 50 mg/ml. To relate our results to a previous study on apoptosis of SMC [32], the cytoki-nes were used at 400 U/ml for TNF-a and IFN-g, and 100 U/ml for Il-1b. In experiments where all three cytokines were used together at these concentrations, this is called the ‘cytokine cocktail’. Alamar blue was purchased from Biosource, and the ApopTag Direct fluorescein in situ apoptosis detection kit was obtained from Oncor.
2.2. Cell culture and 6iability
Human aortic smooth muscle cells and human coro-nary artery endothelial cells (EC) were obtained from Clonetics and were cultured in growth medium from Clonetics with 10% FBS. Cells were seeded at 80% confluency and grown to confluency over a period of 5 days. The cells were not used beyond seven passages. SMC were plated in 96-well plates and cultured with the additions as noted in the text. At the end of the treatment, the medium was removed and replaced with fresh medium including 10% Alamar blue. The cells were then incubated for a further 3 – 4 h and the absorbance was measured at two wavelengths, 570 and 590 nm, in an ELISA plate reader (Molecular Devices Spectra Max 340) and the OD value at 590 nm was subtracted from that at 570 nm. Samples were assayed with eight replicates from each treatment, and the mean OD value calculated.
statin treated cells were incubated with sFasL (100 ng/ml) for 24 h. When added, statins were present throughout the subsequent culture of the cells. After these incubations, the cells were harvested and viability was determined as outlined. Where appropriate, Fas-Ig, mevalonate (500 mmol/l), farnesylpyrophosphate (20 mmol/l) or geranylgeranylpyrophosphate (20 mmol/l), were added to the cultures at the same time as addition of the statins.
2.3. Staining of cells for apoptosis:TUNEL analysis
After the specified treatments, SMC were harvested by collecting both the floating cells by centrifugation and the adherent cells by trypsinization and the popula-tions pooled. Cells were mounted on coverslips, fixed with formalin and the DNA labeled with fluorescein using terminal deoxyribonucleotide transferase accord-ing to the manufacturer’s protocol. Per image, 100 nuclei were counted, and the percentage of TUNEL-positive cells to total number of cells was determined. For each treatment, four different images were ana-lyzed. Error bars indicate the standard deviations.
2.4. Analysis by staining with propidium iodide
SMC were harvested from the tissue culture plates by adding EDTA to the whole culture to a final concentra-tion of 5 mmol/l; plates were incubated for 5 min at 37°C to allow full detachment of SMC. The cells and media were removed, the plates were washed once with RPMI containing 5 mmol/l EDTA and this wash was combined with the original media. Cells were spun down and then permeabilized with buffer containing sodium citrate (0.3%), Triton X-100 (0.01%) and pro-pidium iodide (50 mg/ml). The samples were then ana-lyzed by FACS on a Becton Dickinson FACScan.
3. Results
3.1. Morphological changes in statin-sensitized SMC indicati6e of apoptosis upon treatment with sFasL or cytokines
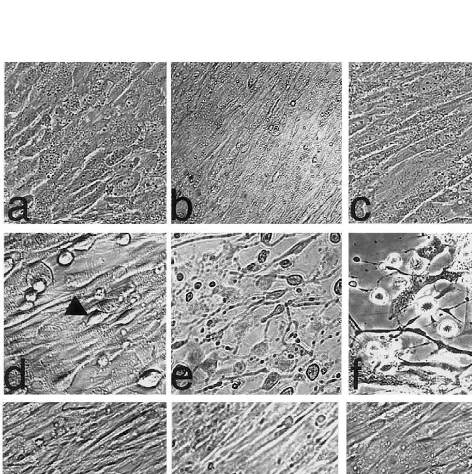
An earlier report on SMC showed that statins at high concentration could induce apoptosis [33], hence initial experiments were carried out to determine whether the statins at low micromolar concentrations would induce visible changes of SMC in culture. Furthermore, since lipophilic statins have been shown to inhibit the prolif-eration of various types of exponentially growing cells [10,34,35], all experiments were performed with conflu-ent cells. Incubation of SMC with simvastatin (3mmol/
l) for 48 or 72 h, induced a slight increase in the number of rounded cells that were detached from the substratum (Fig. 1d) but most of the cells were still adherent. Incubation of SMC without statin pre-treat-ment, but with a cocktail of IFN-g, TNF-a, and Il-1b for 72 h (Fig. 1b), or with sFasL for 24 h (Fig. 1c), did not induce any significant change in the morphology of the cells, in agreement with an earlier report [36]. In contrast, preincubation of the SMC with simvastatin (3 mmol/l) for 24 h followed by stimulation of the cells with the cytokine cocktail for 72 h (Fig. 1e) or sFasL for 24 h (Fig. 1f) resulted in the detachment of many cells from the substratum and prominent shrinkage of the SMC, indicative of cells undergoing apoptosis. Sim-ilar changes were seen upon treatment of SMC with lovastatin (3 mmol/l) and atorvastatin (3 mmol/l), (data not shown). In contrast, pravastatin (20mmol/l) had no effect on its own (Fig. 1g) and did not sensitize SMC to cytokines (Fig. 1h) or sFasL (Fig. 1i).
3.2. Lipophilic statins sensitize SMC to cytokine and sFasL induced apoptosis
The response of SMC to sFasL following statin treatment strongly suggested that the cells were under-going apoptosis. To examine this in more detail, cells Fig. 1. Effect of sFasL or cytokines on the morphology of
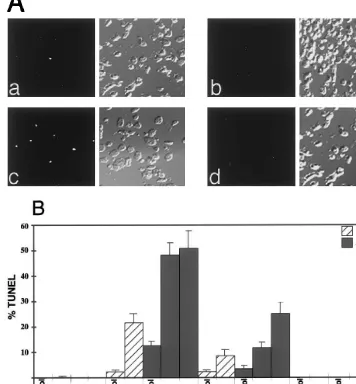
Fig. 2. Induction of TUNEL staining in lipophilic statin-treated cells following stimulation with sFasL or cytokines. Cells were seeded at 96 000 cells/well in six-well plates and allowed to reach confluency. They were then treated with 3mmol/l simvastatin, 6mmol/l atorvastatin, or 5mmol/l pravastatin for 24 h, followed by stimulation with sFasL for a further 24 h. Alternatively cells were treated with 2mmol/l simvastatin, 2mmol/l atorvastatin, or 5mmol/l pravastatin for 24 h, followed by treatment with IFN-galone or the cytokine cocktail for a further 48 h. Cells were fixed with formalin and then stained in situ with fluorescein. Nuclear staining was examined with a fluorescence microscope. (A) Representative TUNEL staining from SMC treated with simvastatin (panel a) or pravastatin (panel b) alone and simvastatin+sFasL (panel c) or pravastatin +sFasL (panel d). Fluorescence and corresponding phase images are shown (original magnification×200). (B) Non-adherent and adherent cells were collected and processed for TUNEL. A total of 100 cells were counted per image and the percentage of TUNEL-positive cells was calculated. For each treatment four different images were analyzed. The mean values (9S.D.) are shown.
were pretreated with statins for 24 h, and then stimu-lated with various combinations of cytokines for an additional 48 h or with sFasL for an additional 24 h. The cells were then analyzed either by FACS staining of nuclei with propidium iodide (Fig. 3) or by TUNEL staining of a pool of both the adherent and floating cells (Fig. 2A and B). Treatment with simvastatin or atorvastatin alone for a total of 48 h induced a low but detectable level of TUNEL positive cells which in-creased slightly over the next 24 h (Fig. 2A and B). Cells that were treated only with cytokines or sFasL were negative for TUNEL staining (Fig. 2B). Addition of sFasL to the simvastatin pretreated SMC cultures
the addition of the cytokine cocktail or sFasL to pravastatin-pretreated SMC (Fig. 2A and B).
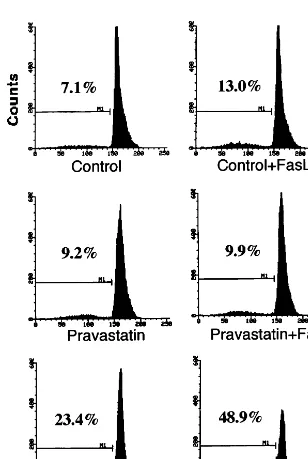
To gain further evidence that SMC were dying by an apoptotic pathway, the ratio of hypodiploid DNA was quantified by propidium iodide staining of cell nuclei (Fig. 3). Cells were treated with statins for 24 h and then stimulated with sFasL for 24 h before they were harvested for analysis by FACS. Simvastatin (2mmol/l) alone increased the content of hypodiploid DNA, whereas pravastatin (5 mmol/l) was without significant effect. sFasL (100 ng/ml) on its own gave rise to a
minor increase in the fragmentation of the DNA, whereas the combination of sFasL with simvastatin pretreatment markedly increased the fragmented DNA. Lovastatin (3 mmol/l), and to a lesser extent atorvas-tatin (5mmol/l), likewise increased the fragmentation of DNA following the addition of sFasL to the SMC cultures (data not shown). In addition, SMC treated with simvastatin (3 mmol/l) and then challenged with sFasL or the cocktail of cytokines showed DNA lad-dering characteristic of programmed cell death (data not shown).
3.3. The sensitization to apoptosis by statins is dose -and cell-type-dependent
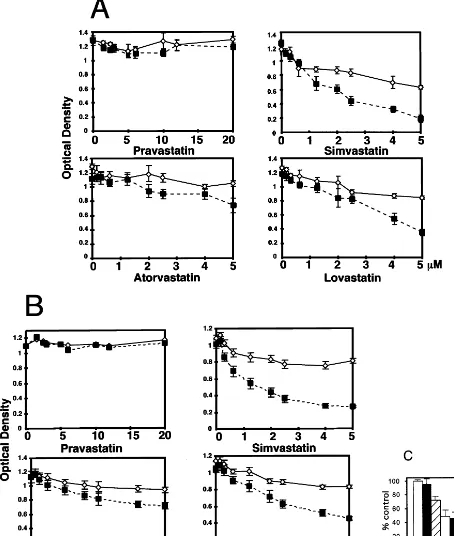
To assess the concentration-dependence of the vari-ous statins in the sensitization of SMC to sFasL or cytokine-induced apoptosis, SMC were incubated with a range of concentrations of the statins for 24 h and then stimulated with the cytokine cocktail for an addi-tional 72 h or with sFasL for 48 h. Cell death was estimated with the dye Alamar blue. Pravastatin, at concentrations up to 20 mmol/l, had no effect on cell viability and did not sensitize the SMC to either sFasL or the cocktail of cytokines (Fig. 4A and B). In con-trast, simvastatin, lovastatin and atorvastatin alone re-duced SMC viability by a small but reproducible degree. Addition of the cytokine cocktail or sFasL to these statin-treated cultures markedly reduced cell vi-ability. The effects of simvastatin, atorvastatin or lovas-tatin on sFasL-induced loss of cell viability could be seen at concentrations down to 150 nmol/l of the statin. Increasing the concentrations of the cytokine cocktail or sFasL above those indicated did not give rise to any additional loss in cell viability (data not shown).
It was of interest to examine if the sensitization of statins was dependent on the cell type. EC were incu-bated with 2mmol/l simvastatin or pravastatin for 24 h and then stimulated with the cytokine cocktail or FasL for an additional 24 h (Fig. 4C). Cell death was as-sessed by Alamar blue staining. In contrast to what we observed with the SMC, the cytokine cocktail alone induced cell death. This was strongly enhanced by further treatment with simvastatin. Further, FasL had no cell death promoting activity in the simvastatin-treated cells.
3.4. Role of Fas in statin-sensitized apoptosis
In order to determine if the enhanced apoptotic sensitivity of statin treated cells was mediated predomi-nantly through the Fas pathway, SMC were incubated with various statins in the absence or presence of 50 mg/ml Fas-Ig. Under these conditions Fas-Ig did not protect the cells from loss of viability due to statin treatment alone (Fig. 5A). In addition, no changes in the levels of cell surface Fas or sFasL could be ob-served following FACS analysis of statin treated cells (data not shown). Fas-Ig was able to block fully the additional cell death that was induced upon addition of sFasL to the statin-treated cell cultures (Fig. 5A) and restored the level of cell viability back to that of cells treated with the statins in the absence of exogenous sFasL.
To determine the role of the Fas pathway in the IFN-g-induction of apoptosis in simvastatin-treated SMC and to characterize whether the sFasL and IFN-g apoptotic pathways were synergistic or overlapping,
cells were sensitized with simvastatin and then treated with a combination of sFasL and IFN-g. sFasL and IFN-g, either alone or in combination, had little effect on cell viability in the absence of statin treatment (Fig. 5B). However, both sFasL and IFN-ginduced a signifi-cant decrease in viability of statin treated cells. The combination of the two agents significantly enhanced SMC death beyond that maximally observed with ei-ther alone. Fas-Ig very effectively reversed the loss in SMC viability in simvastatin-treated cells following treatment with sFasL alone, but did not influence the loss in viability that occurred upon stimulation of statin-treated cells with IFN-galone (Fig. 5B). In addi-tion, Fas-Ig partially reversed the loss in cell viability that occurred following stimulation of simvastatin treated cells with the combination of sFasL and IFN-g. The extent of the inhibition of cell death appeared to be equivalent to that contributed by sFasL. These results suggest that the two pathways leading to apoptosis are distinct but may either additively or synergistically con-tribute to the overall SMC death.
3.5. Role of intermediates in the me6alonate pathway in statin-sensitized apoptosis
Inhibition of HMG CoA reductase within the cell decreases the production of several intermediates in the mevalonate pathway [13], including the generation of farnesylpyrophosphate (FPP), and geranylgeranylpy-rophosphate (GGPP), the substrates for farnesyltrans-ferase and geranylgeranyltransfarnesyltrans-ferase, respectively. The reversibility of the statin-mediated sensitization of SMC to apoptosis was examined by incubation of the cells with simvastatin in the presence of mevalonate, FPP or GGPP for 24 h prior to stimulation with the cytokine cocktail or sFasL. Both mevalonate and GGPP com-pletely blocked the ability of simvastatin to sensitize SMC to either sFasL or cytokine-induced loss of cell viability (Fig. 6). In contrast, FPP gave very little protection suggesting that geranylgeranylation mediates the protection from apoptosis. Similarly both meval-onate and GGPP, but not FPP, protected SMC from apoptosis induced by treatment of the cells with ator-vastatin or loator-vastatin (data not shown).
4. Discussion
Fig. 4. Dose- and cell type-dependent statin sensitization to cytokines and sFasL. SMC and EC were seeded in 96-well plates at 5000 cells/well, grown to confluency and then exposed to various concentrations of statins. After incubation for 24 h the cells were stimulated with (A) the cytokine cocktail for an additional 72 h or (B) sFasL for 48 h. (C) EC were incubated for 24 h with either 2mmol/l simvastatin or 2mmol/l pravastatin and were then stimulated with FasL or the cytokine cocktail for 24 h. Fresh medium containing Alamar blue was added to the cells and the OD determined after 5 h. Optical density (cell viability) is shown for statin treatment alone (open symbol) and statin followed by the cytokines or sFasL (closed symbols). The OD is plotted against statin concentrations. The mean value (9S.D.) was calculated from eight different wells. The values are representative of three independent experiments.
non-lipid lowering effects on extrahepatic tissues is emerging [37]. Distinctions may arise from the ability of the non-hepatic cells to selectively distinguish and
broad range of tissues and cells by passive diffusion [38,39].
Previous studies have shown that several of the lipophilic statins can induce apoptosis in SMC [33] or block activation of T-cells through the T-cell receptor [40]. Many of these studies have been performed at high concentrations of drugs, in some experiments at con-centrations that were 100- to 200-fold above those that
Fig. 6. Effect of mevalonate pathway metabolites on statin-induced sensitization of SMC apoptosis. SMC were seeded at 5000 cells/well in 96-well plates and grown to confluency. SMC were cultured with 3 mmol/l simvastatin in the presence and absence of 500mmol/l meval-onate (M), geranylgeranylpyrophosphate (GGPP) or farnesylpy-rophosphate (FPP) for 24 h. The cytokine cocktail or sFasL was added to the wells and the cells incubated for an additional 48 h. The viability of the cells was then determined with Alamar blue. The optical density was determined 5 h after the addition of Alamar blue. The mean value of the optical density (9S.D.) derived from eight replicate wells per sample was determined. The values are representa-tive of three independent experiments.
Fig. 5. Role of Fas in FasL and cytokine-induced apoptosis. SMC were seeded in 96-well plates at 5000 cells/well and grown to conflu-ency prior to initiation of treatments. (A) Cells were incubated with 2 mmol/l of either simvastatin, atorvastatin, or lovastatin together with Fas-Ig for 24 h, prior to the addition of sFasL. The 10mmol/l pravastatin was not different from control (data not shown). Cells were then incubated for a further 48 h and viability determined by the addition of Alamar blue. (B) SMC were cultured with 3 mmol/l simvastatin for 24 h in the presence or absence of Fas-Ig. The cells were then treated for an additional 48 h with IFN-gor sFasL alone or in combination. Cell viability was then determined by the addition of Alamar blue and the optical density was determined after 5 h. The mean value of the optical density (9S.D.) derived from eight repli-cate wells per sample was determined. The values are representative of three independent experiments.
are achieved in the plasma [33] and in species other than human [35]. Thus, the relevance to the physiologi-cal situation is unclear.
In this study we have examined the effect of the different statins on human SMC physiology at low doses. At low concentrations, only the lipophilic statins had a low but detectable effect on apoptosis and cell viability of the SMC. Simvastatin was the most potent, followed by lovastatin and atorvastatin. On the other hand, the hydrophilic pravastatin did not induce any detectable cell death at concentrations up to 20mmol/l. Since some of the statins are subject to drug interac-tions with CYP3A4 inhibitors, which can lead to in-creases in plasma levels by as much as 19-fold to a serum concentration of 2 mmol/l [41], the levels studied are especially relevant. Similar levels are reached with grapefruit juice [42]. Even though in the experiments reported here, the effects were seen at submicromolar concentrations of the statins, it is un-clear whether the levels of statins achieved in vivo in the tissues would reach those that appear to be neces-sary to induce sensitization in vitro. However, tissue levels may be higher and duration of exposure longer than in the plasma [43] and repeated exposure may alter the sensitivity of the cells.
apop-totic response in human SMC [36]. Similar to what was reported [20], we have found that cytokines alone are unable to sensitize the cells to FasL induced apoptosis. The differences in reports may be due to different cell culture conditions. We show here that pretreatment of SMC with low doses of lipophilic statins renders the cells susceptible to the induction of programmed cell death by these agents. Taken to-gether, the results from the three groups indicate that the inflammatory environment plays a major role in SMC survival. In contrast to the SMC, we found that EC pretreated with statins undergo apoptosis follow-ing stimulation with cytokines but are resistant to sFasL. Thus, the resistance to Fas mediated cell death in EC as also reported by others [44,45] cannot be overcome by treatment with statins.
Statin-treatment sensitized SMC to both cytokines and sFasL. The intracellular pathways leading to cell death appear to be different but cumulative. This is supported by several observations. For example, Fas-Ig was able to block sFasL-induced death but not the loss in cell viability induced by higher concentrations of statins alone or that induced by cytokines in the presence of statins. Furthermore, treatment of statin-sensitized SMC with a combination of sFasL and the cytokines induced higher levels of apoptosis than that seen maximally with either treatment alone. Finally, a combination of caspase inhibitors, Ac-YVAD-CHO, Z-VAD-FMK, and Z-DEVD-FMK, that have been shown previously to block Fas-induced cell death [46], inhibited sFasL-induced death but not that induced by the statins or the cytokines alone (data not shown).
The mechanisms by which the statins induce sensi-tization to subsequent apoptotic challenge are not fully understood. In SMC that had been treated with simvastatin, lovastatin or atorvastatin no differences in the cell surface expression of Fas or sFasL could be detected (data not shown). This suggests that nei-ther the statin-induced spontaneous cell death nor the enhanced response to sFasL can be attributed simply to an increase in the expression of these apoptotic mediators.
Mevalonate and GGPP, but not FPP, abrogated
the sensitization of SMC to apoptosis by the
lipophilic statins. Many proteins are differentially far-nesylated and geranylgeranylated including Ras, nu-clear lamin and mammalian G proteins. Inhibition of prenylation alters many cellular functions [47]. We show here that inhibition of geranylgeranylation, but not farnesylation enhances apoptosis in pulmonary vascular SMC, which is consistent with a recent re-port [48]. Thus, it appears that geranylgeranylated proteins protect SMC against apoptosis. However, the target proteins are as yet undefined. Recently it has been shown that both retrograde and anterograde
transport (e.g. of Golgi glycosyltransferases) between the Golgi apparatus and the endoplasmatic reticulum involves prenylated proteins [49], which could be blocked by lovastatin. In one study, activation of p53 transiently increased transport of Fas from the Golgi to the cell surface and also enhanced sensitivity of cells to sFasL by the induction of Fas-FADD binding [50]. In contrast, in our studies the statins did not increase cell surface expression of Fas (data not shown). Furthermore, Ig, an inhibitor of the Fas-FasL interaction, did not prevent the small but sig-nificant level of apoptosis induced by statins alone or the much greater cytokine-induced apoptosis in statin-sensitized cells. Thus, it seems unlikely that increased cell surface expression of Fas can solely account for the statin induced sensitization of SMC to apoptosis. Rather, geranylgeranylated proteins may be involved in regulating the functional assembly of an intracellu-lar death inducing signaling complex.
The in vivo effect of the different statins on the apoptosis of SMC and other cells or tissues may be difficult to demonstrate. However, several studies have shown that in the absence of changes in lipid serum levels, lipophilic statins inhibit SMC growth in vivo [6,51 – 54]. Also, Corsini et al. found ex vivo evi-dence that plasma from fluvastattreated patients in-hibited SMC proliferation in contrast to that from pravastatin-treated patients [34]. Additionally, SMC in the atherosclerotic intima seem to be much more susceptible to apoptosis [55,56].
In summary, our results demonstrate that low con-centrations of lipophilic statins sensitize SMC to apoptosis. One hypothesis predicts that at sites of plaque thinning, death of SMC will decrease plaque stability, which could give rise to increased plaque vulnerability, rupture and possibly a fatal thrombosis [5]. Thus, under suitable conditions (e.g. local inflam-mation), by diminishing the number of SMC, the lipophilic statins could enhance plaque thinning. On the contrary, in restenosis this effect could be benefi-cial. The effect in vivo may vary according to the stage of the disease. Furthermore, lipophilic statins may well sensitize other cells within the plaque or at completely different locations to apoptosis. The physi-ological significance of this sensitization, not only to cardiovascular disease, but also to other disease states such as inflammatory disorders remains to be deter-mined.
Acknowledgements
References
[1] Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. New Engl J Med 1996;335:1001 – 9.
[2] The Scandinavian Simvastatin Survival Study. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease (4S). Lancet 1994;344:1383 – 9.
[3] Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFar-lane PW, et al. Prevention of coronary heart disease with pravas-tatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. New Engl J Med 1995;333:1301 – 7.
[4] Ross R. The pathogenesis of atherosclerosis: perspective for the 1990s. Nature 362;1993:801 – 9.
[5] Libby P, Geng Y-J, Sukhova GK, Simon DI, Lee RT. Molecular determinants of atherosclerotic plaque vulnerability. Ann NY Acad Sci 1997;811:134 – 45.
[6] Bellosta S, Bernini F, Ferri N, Quarato P, Canavesi M, Arn-aboldi L, et al. Direct vascular effects of HMG-CoA reductase inhibitors. Atherosclerosis 1998;137:S101 – 9.
[7] Massy ZA, Keane WF, Kasiske BL. Inhibition of the meval-onate pathway: benefits beyond cholesterol reduction? Lancet 1996;347:102 – 3.
[8] West of Scotland Coronary Prevention Study Group. Influence of pravastatin and plasma lipids on clinical events in the West of Scotland Coronary Prevention Study [WOSCOPS]. Circulation 1998;97:1440 – 5.
[9] Kiener PA, Davis PM, Murray L, Youssef S, Rankin BM, Kowala M. Characterization of the differences in the pro-inflam-matory effects of HMG CoA reductase inhibitors. In: European Atherosclerosis Society 70th EAS Congress, Geneva, Switzer-land, September 6 – 9, 1998, 1998 (abstract).
[10] Negre-Aminou P, van Vliet AK, van Erck M, van Thiel GCF, van Leeuwen REW, Cohen LH. Inhibition of proliferation of human smooth muscle cells by various HMG-CoA reductase inhibitors: comparison with other human cell types. Biochem Biophys Acta 1997;1345:259 – 68.
[11] Colli S, Eligini S, Lalli M, Camera M, Paoletti R, Tremoli E. Vastatins inhibit tissue factor in cultured human macrophages. A novel mechanism of protection against atherothrombosis. Arte-rioscler Thromb Vasc Biol 1997;17:265 – 72.
[12] Pirillo A, Jacoviello C, Longoni C, Radaelli A, Catapano AL. Simvastatin modulates the heat shock response and cytotoxicity mediated by oxidized LDL in cultured human endothelial smooth muscle cells. Biochem Biophys Res Commun 1997;231:437 – 41.
[13] Goldstein JL, Brown MS. Regulation of the mevalonate path-way. Nature 1990;343:425 – 30.
[14] Libby P, Ross R. Cytokines and growth regulatory molecules in atherosclerosis. In: Fuster V, Ross R, Topol EJ, editors. Atherosclerosis and coronary artery disease. Philadelphia, PA: Lippincott-Raven, 1996:585 – 94.
[15] Libby P. Atheroma: more than mush. Lancet 1996;348:S4 – 7. [16] DeGraba TJ. Expression of inflammatory mediators and adhe-sion molecules in human atherosclerotic plaque. Neurology 1997;49:S15 – 9.
[17] Buja LM, Willerson JT. Role of inflammation in coronary plaque disruption. Circulation 1994;89:503 – 5.
[18] Van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atheroscle-rotic plaques is characterized by an inflammatory process irre-spective of the dominant plaque morphology. Circulation 1994;89:36 – 44.
[19] Kockx MM, Herman AG. Apoptosis in atherogenesis: implica-tions for plaque destabilization. Eur Heart J 1998;19:G23 – 8. [20] Geng Y-J, Henderson LE, Levesque EB, Muszynski M, Libby P.
Fas is expressed in human atherosclerotic intima and promotes apoptosis of cytokine-primed human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 1997;17:2200 – 8.
[21] Cai W-j, Devaux B, Schaper W, Schaper J. The role of Fas/APO 1 and apoptosis in the development of human atherosclerotic lesions. Atherosclerosis 1997;131:177 – 86.
[22] Crisby M, Kallin B, Thyberg J, Zhivotovsky B, Orrenius S, Kostulas V, et al. Cell death in human atherosclerotic plaques involves both oncosis and apoptosis. Atherosclerosis 1997;130:17 – 27.
[23] Bennett MR, Boyle JJ. Apoptosis of vascular smooth muscle cells in atherosclerosis. Atherosclerosis 1998;138:3 – 9.
[24] Schwartz SM, deBlois D, O’Brien ERM. The intima. Soil for atherosclerosis and restenosis. Circ Res 1995;77:445 – 65. [25] Geng Y-J, Libby P. Evidence for apoptosis in advanced human
atheroma. Colocalization with interleukin-1b-converting enzyme. Am J Pathol 1995;147:251 – 66.
[26] Kockx MM, DeMeyer GRY, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation 1998;97:2307 – 15. [27] Bennett M, Macdonald K, Chan S-W, Boyle JJ, Weissberg PL. Cooperative interactions between RB and p53 regulate cell pro-liferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res 1998;82:704 – 12.
[28] Perlman H, Maillard L, Krasinski K, Walsh K. Evidence for the rapid onset of apoptosis in medial smooth muscle cells after balloon injury. Circulation 1997;95:981 – 7.
[29] Geng Y-J. Regulation of programmed cell death or apoptosis in atherosclerosis. Heart Vessels 1997;12:76 – 80.
[30] Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase in-hibitors. Circulation 1998;97:1129 – 35.
[31] Kiener PA, Davis PM, Starling GC, Mehlin C, Klebanoff SJ, Ledbetter JA, et al. Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J Exp Med 1996;185:1511 – 6.
[32] Geng Y-J, Wu Q, Muszynski M, Hansson GK, Libby P. Apop-tosis of vascular smooth muscle cells induced by in vitro stimula-tion with interferon-g, tumor necrosis factor-a and interleukin-1b. Arterioscler Thromb Vasc Biol 1996;16:19 – 27. [33] Guijarro C, Blanco-Colio LM, Ortego M, Alonso C, Ortiz A,
Plaza JJ, et al. 3-Hydroxy-3-methylglutaryl Coenzyme A reduc-tase and isoprenylation inhibitors induce apoptosis of vascular smooth muscle cells in culture. Circ Res 1998;83:490 – 500. [34] Corsini A, Pazzucconi F, Arnaboldi F, Pfister L, Fumagalli R,
Paoletti R, et al. Direct effects of statins on the vascular wall. J Cardiovasc Pharmacol 1998;31:773 – 8.
[35] Baetta R, Donetti E, Comparato C, Calore M, Rossi A, Teruzzi C, et al. Proapoptotic effect of atorvastatin on stimulated rabbit smooth muscle cells. Pharmacol Res 1997;36:115 – 21.
[36] Jovinge S, Crisby M, Thyberg J, Nilsson J. DNA fragmentation and ultrastructural changes of degenerating cells in atheroscle-rotic lesions and smooth muscle cells exposed to oxidized LDL in vitro. Arterioscler Thromb Vasc Biol 1997;17:2225 – 31. [37] Rosenson RS, Tangney CC. Antiatherothrombotic properties of
statins: implications for cardiovascular event reduction. J Am Med Assoc 1998;279:1643 – 50.
[39] Hamelin BA, Turgeon J. Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reduc-tase inhibitors. Trends Pharmacol Sci 1998;19:26 – 37.
[40] Goldman F, Hohl RJ, Crabtree J, Lewis-Tibesar K, Koretzky G. Lovastatin inhibits T cell antigen receptor signaling independent of its effects on ras. Blood 1996;88:4611 – 9.
[41] Neuvonen PJ, Kantola T, Kivistoe KT. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther 1998;63:332 – 41. [42] Lilja JJ, Kivisto KT, Neuvonen PJ. Grapefruit juice-simvastatin
interaction: effect on serum concentrations of simvastatin, sim-vastatin acid, and HMG-CoA reductase inhibitors. Clin Pharma-col Ther 1998;64:477 – 83.
[43] Duggan DE, Chen I-W, Bayne WF, Halpin RA, Duncan CA, Schwartz MS, et al. The physiological disposition of lovastatin. Drug Metab Dispos 1989;17:166 – 73.
[44] Sata M, Walsh K. TNFaregulation of Fas ligand expression on vascular endothelium modulates leukocyte extravasation. Nat Med 1998;4:415 – 20.
[45] Richardson BC, Lalwani ND, Johnson KJ, Marks RM. Fas ligation triggers apoptosis in macrophages but not endothelial cells. Eur J Immunol 1994;24:2640 – 5.
[46] Longthorne VL, Williams GT. Caspase activity is required for commitment to Fas-mediated apoptosis. EMBO J 1997;13:3805 – 12.
[47] Tamanoi F. Inhibition of Ras farnesyltransferases. Trends Biochem Sci 1993;18:349 – 53.
[48] Stark JR WW, Blaskovich MA, Johnson BA, Qian Y, Vasude-van A, Pitt B, et al. Inhibiting geranylgeranylation blocks growth and promotes apoptosis in pulmonary vascular smooth muscle cells. Am J Physiol 1998;275:L55 – 63.
[49] Ivessa NE, Gravotta D, De Lemos-Chiarandini C, Kreibich G. Functional protein prenylation is required for the brefeldin A-dependent retrograde transport from the Golgi apparatus to the endoplasmic reticulum. J Biol Chem 1997;272:20828 – 34. [50] Bennett M, Macdonald K, Chan S-W, Luzio JP, Simari R,
Weissberg P. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science 1998;282:290 – 3.
[51] Igarashi M, Takeda Y, Mori S, Ishibashi N, Komatsu E, Taka-hashi K, et al. Suppression of neointimal thickening by a newly developed HMG-CoA reductase inhibitor, BAYw6228 and its inhibitory effect on vascular smooth muscle cell growth. Br J Pharmacol 1997;120:1172 – 8.
[52] Bandoh T, Mitani H, Niihashi M, Kusumi Y, Ishikawa J, Kimura M, et al. Inhibitory effect of fluvastatin at doses insuffi-cient to lower serum lipids on the catheter-induced thickening of intima in rabbit femoral artery. Eur J Pharmacol 1996;315:37 – 42.
[53] Soma M, Donetti E, Parolini C, Mazzini G, Ferrari C, Fuma-galli R, et al. HMG-CoA reductase inhibitors. In vivo effects on carotid intimal thickening in normocholesterolemic rabbits. Ar-terioscler Thromb 1993;13:571 – 8.
[54] Gellman J, Ezekowitz MD, Sarembock IJ, Azrin MA, Nocho-mowitz LE, Lerner E, et al. Effect of lovastatin on intimal hyperplasia after balloon angioplasty: a study in an atheroscle-rotic hypercholesterolemic rabbit. J Am Coll Cardiol 1991;17:251 – 9.
[55] Mitchinson MJ, Hardwick SJ, Bennett MR. Cell death in atherosclerotic plaques. Curr Opin Lipidol 1996;7:324 – 9. [56] Bennett MR, Evan GI, Schwartz SM. Apoptosis of human
smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest 1995;95:2266 – 74.