Synthesis of N-(phenylcarbamothioyl)-benzamide derivatives and their cytotoxic activity against MCF-7 cells - Ubaya Repository
Teks penuh
Gambar
Dokumen terkait
menciptakan kondisi belajar bagi peserta didik. Rumusan ini mengandung konsep sebagai berikut: 1) Pembelajaran bertujuan mengembangakan atau mengubah tingkah laku peserta didik,
Membaca buku sambil tiduran menyebabkan mata menjadi minus karena posisi tidak ergonomis, maka jarak membaca tidak ideal. Seharusnya jarak membaca adalah lebih
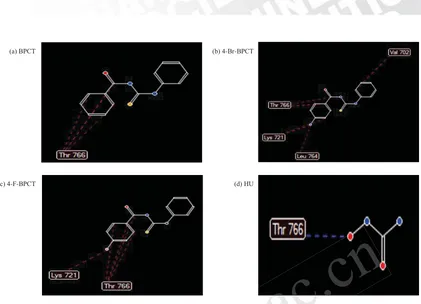
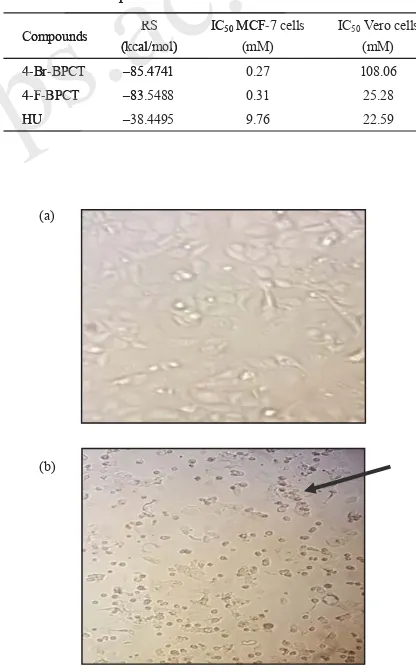
Based on the results, the cytotoxic activity value of the steroids 1-7 against MCF-7 breast cancer cells is affected by the skeleton type of steroids and changes in substituents
This supplementary data is a part of a paper entitled “Steroids from Atactodea striata and Their Cytotoxic Activity against MCF-7 Breast Cancer Cell Lines”.. Contents of
leaves ethanol extracts, fractions, and main compounds against breast cancer cells (MCF-7).. Cytotoxic activity was carried out by the
Tanggal atas efektifnya penggabungan 30 September 2011 Tanggal Pembayaran atas pembelian saham dari pemegang saham 03 Oktober 2011 publik yang telah menyatakan maksud mereka
Berdasarkan penelitian yang dilakukan mahasiswa memiliki motivasi belajar dengan baik dengan predikat prestasi belajar memuaskan (2,00-2,75) sebanyak 2 mahasiswa
Berdasarkan hasil implementasi Model Audit Pertanggungjawaban Sosial berbasis HCD ditemu- kan adanya beberapa kondisi atas organisasi sektor publik yang diaudit, diantaranya
