BAB I PENDAHULUAN 1.1Latar Belakang
Sterilisasi adalah cara untuk mendapatkan suatu kondisi bebas mikroba atau setiap proses yang dilakukan baik secara fisika, kimia, dan mekanik untuk membunuh semua bentuk kehidupan terutama mikroorganisme. Dalam bidang mikrobiologi baik dalam pengerjaan penelitian atau praktikum, keadaan steril merupakan syarat utama berhasil atau tidaknya pekerjaan kita dilaboratorium.Pengetahuan tentang prinsip dasar sterilisasi dan desinfeksi sangat diperlukan untuk melakukan pekerjaan di bidang medis yang bertanggung jawab.Setiap proses (baik fisika, kimia maupun mekanik) yang membunuh semua bentuk kehidupan terutama mikroorganisme disebut dengan sterilisasi. Adanya pertumbuhan mikroorganisme menunjukkan bahwa pertumbuhan bakteri masih berlangsung dan tidak sempurnanya proses sterilisasi. Jika sterilisasi berlangsung sempurna, maka spora bakteri yang merupakan bentuk paling resisten dari kehidupan mikroba.
Pada preparasi sediaan steril, produk steril haruslah dibuat dengan persyaratan khusus, dengan tujuan meniadakan (memperkecil) resiko kontaminasi mikroba, partikel partikulat, pirogen, dan produk interaksi lainnya (misal dengan kemasan dan penutup kemasan); yang sangat tergantung pada derajat kemurnian bahan baku dan tergantung pula pada keterampilan, tanggung jawab, dan sikap operator yang terlibat selama proses produksi. Jaminan dan pemastian mutu penting sekali artinya karena hampir tidak mungkin produk steril menghadapi proses ulang (rework). Oleh karena itu, cara (proses) pembuatan haruslah mengikuti prosedur yang divalidasi.
Proses pembuatan produk farmasi sediaan steril perlu memperhatikan proses sterilisasi produk untuk menghindari terjadinya kontaminasi pada sediaan steril yang akan diproduksi dimana dapat berdampak pada pengguna sediaan steril tersebut sehingga perlu dilakukan proses yang sangat ketat dalam setiap tahapan pengolahannya. Salah satu upaya yang dilakukan yakni melakukan proses produksi sediaan steril dengan menggunakan 2 metode yakni proses aseptis dan
proses sterilisasi akhir. Dimana perlu diketahui tahapan dari setiap metode tersebut agar menghasilkan sediaan farmasi steril yang bermutu, aman dan bebas dari mikroba dan pirogen untuk masyarakat yang menggunakannya.
1.2 Rumusan Masalah
1. Bagaimana tahapan proses produksi secara aseptis dan sterilisasi akhir ? 2. Bagaimana proses pengolahan produk steril ?
3. Bagaimana proses sterilisasi produk steril ?
4. Bagaimana tahapan proses produksi produk steril padat, semipadat, dan cair ?
5. Bagaimana proses pengawasan mutu dari produk steril ? 1.3 Tujuan
1. Mengetahui tahapan proses produksi secara aseptis dan sterilisasi akhir 2. Mengetahui proses pengolahan produk steril
3. Mengetahui cara sterilisasi produk steril
4. Mengetahu tahapan proses produksi steril padat, semipadat dan cair 5. Mengetahui proses pengawasan mutu dari produk steril
BAB II ISI 1.1 Pembuatan secara aseptis
a. Definisi proses aseptis
Proses aseptis adalah metode pembuatan produk steril menggunakan saringan dengan filter khusus untuk bahan obat steril atau bahan baku steril yang diformulasikan dan diisikan kedalam container steril serta dilakukan dilingkungan terkontrol. Suplai udara, material, peralatan, dan petugas telah terkontrol sedemikian rupa sehingga kontaminasi mikroba tetap berada pada level yang dapat diterima yakni dalam area bersih (grade A dan B). persyaratannya adalah limit of media fill 1:10.000 unit dapat dikatakan produk beas mikroorganisme. Proses demikian dipilih bila obat atau bahan obat yang akan diproduksi tidak tahan panas (lukas, 2011).
b. Tujuan dari proses aseptis adalah untuk mempertahankan sterilitas produk yang dibuat darikomponen-komponen yang masing-masing telah disterilisasi sebelumnya dengan menggunakan salah satu cara dari metode yang ada. Kondisi operasional hendaklah dapat mencegah kontaminasi mikroba. Untuk menjaga sterilitas komponen dan produk selama proses aseptis, perhatian perlu diberikan pada :
1. Lingkungan 2. Personil
3. permukaan yang kritis;
4. sterilisasi wadah/ tutup dan prosedur pemindahannya;
5. waktu tunggu maksimum bagi produk sebelum pengisian ke dalam wadah akhir
6. filter untuk sterilisasi.
c. Langkah-langkah proses aseptis pada produksi steril
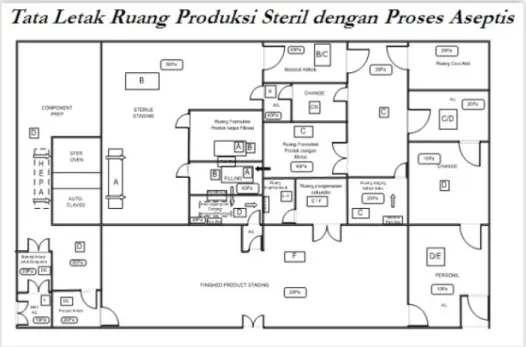
1. Bahan yang telah dicuci ditangani di lingkungan minimal Kelas D. Penanganan bahan awal dan komponen steril, kecuali pada proses selanjutnya untuk disterilisasi atau disaring dengan menggunakan filter mikroba dilakukan di lingkungan Kelas A dengan latar belakang Kelas B. Untuk produk yang berisiko besar terhadap kontaminasi
partikel selama proses, misalnya infus bervolume >100ml, dan produk dalam wadah bermulut lebar maka pembilasan akhir dan penanganan komponen setelah dicuci hendaklah dilakukan di bawah LAF yang dipasang di lingkungan minimal Kelas D.
Gambar 1. Proses produksi steril secara aseptis (CPOB, 2012)
2. Proses pembuatan larutan yang akan disterilisasi secara filtrasi dilakukan di lingkungan Kelas C; bila tidak dilakukan filtrasi, penyiapan bahan dan produk hendaklah dilakukan di lingkungan Kelas A dengan latar belakang Kelas B.
3. Penanganan dan pengisian produk yang dibuat secara aseptis hendaklah dilakukan di lingkungan Kelas A dengan latar belakang Kelas B.
4. Transfer wadah setengah-tertutup, yang akan digunakan dalam proses beku-kering (freeze drying) hendaklah, sebelum proses penutupan dengan stopper selesai, dilakukan di lingkungan Kelas A dengan latar belakang Kelas B atau dalam nampan transfer yang tertutup di lingkungan Kelas B.
5. Pembuatan dan pengisian salep, krim, suspensi dan emulsi hendaklah dilakukan di lingkungan Kelas A dengan latar belakang Kelas B, apabila produk terpapar dan tidak akan disaring.
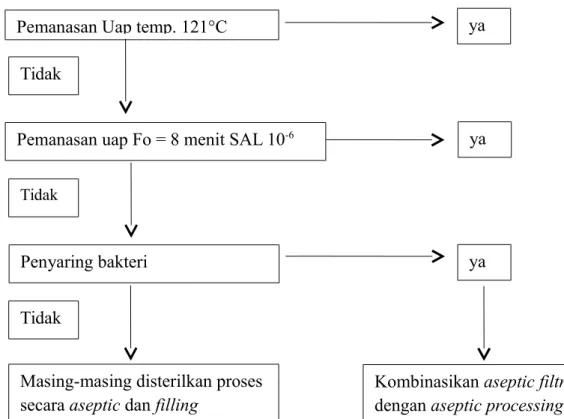
Beberapa pilihan menurut Current Pharmaceutical Manufacturing Practice (CPMP) USA FDA (2004) adalah produk dapat disterilkan dengan beberapa cara seperti gambar berikut dibawah ini
Gambar 2. Alur pemilihan cara sterilisasi (Lukas, 2011) 1.2 Pembuatan produk yang disterilisasi akhir
Penyiapan komponen dan sebagian besar produk yang memungkinkan untuk disaring dan disterilisasi harus dilakukan minimal di lingkungan kelas D untuk mengurangi resiko cemaran mikroba dan cemaran partikel partikulat. Bila ada resiko terhadap produk akibat cemaran mikroba, misalnya produk yang secara aktif mendukung pertumbuhan mikroba atau harus didiamkan selama beberapa saat sebelum sterilisasi atau terpaksa diproses dalam tangki tidak tertutup maka penyiapan hendaklah dilakukan di lingkungan kelas C.
Pemanasan Uap temp. 121°C ya
Tidak
Pemanasan uap Fo = 8 menit SAL 10-6 ya
Tidak
ya Penyaring bakteri
Tidak
Kombinasikan aseptic filtration dengan aseptic processing Masing-masing disterilkan proses
Pengisian produk yang akan disterilkan dengan cara sterilisasi akhir harus dilakukan di lingkungan minimal kelas C. Bila ada resiko terhadap produk karena cemaran lingkungan, misalnya karena kegiatan pengisian berjalan lambat atau wadah berleher lebar atau terpaksa terpapar lebih dari beberapa detik sebelum ditutup, pengisian hendaklah dilakukan di zona kelas A dengan latar belakang minimal kelas C. Penyiapan dan pengisian salep, krim, suspensi, dan emulsi pada umumnya hendaklah dilaksanakan di lingkungan kelas C sebelum dilakukan sterilisasi akhir.
Metode sterilisasi akhir menurut PDA Technical Monograph (2005) dibagi menjadi dua :
1. Overkill methode adalah metode sterilisasi menggunakan pemanasan dengan uap panas pada suhu 121°C selama 15 menit yang mampu memberikan minimal reduksi setingkat log 12 dari berbagai mikroorganisme yang memiliki nilai D minimal 1 menit. Kriteria sterilitas yang digunakan adalah probabilitas survival tidak lebih besar dari satu mikroorganisme dalam 106 unit. Metode ini digunakan untuk bahan yang tahan panas seperti zat anorganik. Metode ini merupakan pilihan utama karena lebih efisien, cepat, dan aman.
2. Bioburden sterilization adalah metode sterilisasi yang memerlukan monitoring lengkap dan terkontrol terhadap beban mikroba sekecil mungkin di beberapa lokasi jalur produksi sebelum menjalani proses sterilisasi lanjutan dengan tingkat sterilitas yang dipersyaratkan SAL 106. Metode ini digunakan untuk bahan yang dapat mengalami degradasi kandungan apabila dipanaskan terlalu tinggi, seperti zat organik.
Perbedaan kedua metode tersebut adalah titik awal. Apabila menggunakan pendekatan overkill maka pemanasan dilakukan dengan uap pada suhu 121°C selama 15 menit, sedangkan pendekatan bioburden dilihat dari pencapaian tingkat sterilitas yang diminta, yaitu SAL 10-6.
1.3 Teknologi Isolator
Isolator dapat didefinisikan sebagai suatu alat yang menyediakan kondisi tertutup terkendali atau lingkungan bersih dimana dilakukan suatu
proses atau aktivitas yang dapat menjamin bahwa pemisahan secara efektif dapat dipertahankan antara lingkungan tertutup, lingkungan sekitarnya dan setiap personalia yang terlibat dalam proses. Isolator dapat didesain secara tertutup atau terbuka, dan dapat mempertahankan tekanan udara positif atau negatif terhadap lingkungan sekitar.
Penggunaan isolator terutama bertujuan untuk meningkatkan integritas proses secara menyeluruh. Dalam hal ini termasuk perlindungan terhadap operator dari material poten dan berbahaya, sedangkan pada manufaktur produk steril untuk mengurangi potensial kontaminasi unit nonsteril yang dihasilkan dari proses spesifik. Selain itu isolator dalam beberapa hal digunakan untuk meminimalkan pemakaian lokasi ruangan (space) dan meminimalkan biaya operasional.
1.4 Teknologi peniupan/pengisian/penyegelan
Pada saat ini teknologi peniupan/pengisian/penyegelan terutama dikembangkan untuk produk farmasi steril, seperti larutan respiratori (untuk dihirup), obat mata, produk perawatan luka. Teknologi peniupan/pengisian/penyegelan adalah teknik aseptik lanjutan (advance) dimana kontener plastik dibentuk melalui cara ekstrusi penuangan granul polimer yang diisikan dan disegel melalui suatu proses kontinu. Hal ini berbeda dari proses aseptik konvensional dimana pembentukan kontener, preparasi, sterilisasi, dan pengisian serta penutupan kontener semuanya dilakukan terpisah.
Akibat tingkat otomatisasi dari keseluruhan proses, teknologi ini sedikit sekali memerlukan intervensi manusia selama proses manufaktur jika dibandingkan dengan proses aseptik secara tradisional. Hal ini dianggap sebagai pengisian proses aseptik lebih lanjut (advance). Oleh sebab itu, dengan menggunakan teknologi aseptik secara sempurna, akan dapat dicapai tingkat sterilitas yang tinggi.
Mesin peniup/pengisi/penyegel me-rupakan satu rangkaian mesin, di mana, dalam suatu operasi yang kontinu, wadah produk dibentuk dari granulat termoplastis, diisi dan kemudian disegel, semua ini dilakukan oleh satu unit mesin otomatis. Mesin peniup/pengisi/penyegel yang digunakan
untuk produksi aseptis yang dilengkapi dengan air shower yang efektivitasnya sama dengan Kelas A dapat dipasang dalam lingkungan minimal Kelas C, dengan syarat mengenakan pakaian kerja Kelas A/B. Mesin yang digunakan untuk pembuatan produk dengan sterilisasi akhir hendaklah dipasang dalam lingkungan minimal Kelas D.
Lingkungan kerja hendaklah memenuhi persyaratan jumlah partikel dan mikroba pada kondisi “nonoperasional” dan persyaratan jumlah mikroba hanya pada saat beroperasi.
1.5 Air
1. Pengertian
Air merupakan bahan awal yang sangat penting, maka mutunya harus dikendalikan yang dimulai dengan kualifikasi kinerja Sistem Pengolahan Air(Water Treatment System).Air yang dipakai untuk membuat produk steril termasuk penyimpanan dan sistem distribusinya harus selalu dikendalikan untuk menjamin bahwa spesifikasi yang sesuai dicapai tiap pengoperasian.Air yang digunakan untuk formulasi hendaklah diperlakukan sebagai bahan awal. Maksudnya yakni dilakukan pemisahan secara fisik atau cara lain yang tervalidasi. Kemudian dilakukan penyimpanan bahan yang ditolak maupun yang diterima dengan memberikan identitas yang tepat.Kemudian diserahkan kebagian area penyimpanan untuk diperiksa kebenaran identitas, kondisi wadah dan tanda pelulusan oleh bagian Quality control. Jika tidak sesuai dengan mutu maka akan dikirim kebagian karantina dan akan ditentukan statusnya oleh bagian QC terkait status bahan tersebut. Pada bahan awal diberlakukan system FIFO dan FEFO kemudian dilakukan uji ulang pada bahan yang telah lama disimpan untuk menjamin mutu bahan awal tersebut (Lukas, 2011)
Air untuk Injeksi (WFI) diproduksi melalui cara penyulingan atau cara lain yang akan menghasilkan mutu yang sama. Air untuk Injeksi (WFI) diproduksi, disimpan dan didistribusikan dengan cara yang dapat mencegah pertumbuhan mikroba, misal disirkulasi dengan konstan pada suhu di atas 70°C atau tidak lebih dari 4°C, bila air untuk injeksi
disirkulasikan, hendaklah dibuang setelah 24 jam. Air untuk Injeksi (WFI) disimpan dalam wadah yang bersih, steril, nonreaktif, nonabsorptif, nonaditif dan terlindung dari pencemaran. (CPOB, 2012) Sumber air, peralatan pengolahan air dan air hasil pengolahan hendaklah dipantau secara teratur terhadap pencemaran kimiawi, biologis dan, bila perlu, terhadap cemaran endotoksin untuk menjamin agar air memenuhi spesifikasi yang sesuai dengan peruntukannya. Hasil pemantauan dan tindakan penanggulangan yang dilakukan hendaklah didokumentasikan.Alat perekam hendaklah digunakan untuk memantau suhu penyimpanan (CPOB, 2012)
2. Proses Pembuatan Air Untuk Injeksi
a. Proses pertama adalah persiapan (pretreatment) untuk mendapatkan water For Injection dimulai dari sumber air (sumur atau mata air) yang ditampung dan diendapkan. Kemudian diberi penyaring dan diberi klorin sehingga air dapat diminum (drinking water). Air minum disaring dengan karbonaktif, lalu disaring kembali dengan filter 5-10 µm.
b. Proses kedua adalah final treatment, biasanya dilakukan dengan reverse osmosis dengan menggunakan chemical softening (kation dan anion) atau menggunakan twin bed column, lalu disaring menggunakan filter 5-10µm. selanjutnya disaring lagi mengguakan filter yang lebih kecil dengan ukuran 2 µm, bila perlu menggunakan ozonisator atau ultraviolet atau pemanasan dengan temperature diatas 70°C, kemudian dimasukkan kedalam tangki penampung dengan temperature 70°C . setelah itu di EDI (Electro Deionization) atau didestilasi dimasukkan kedalam tangki penampung, lalu disaring dengan filter bakteri 0,2µm
c. Proses ketiga adalah sterilisasi WFI dengan menggunakan autoklaf sehingga mendapatkan WFI steril (Lukas, 2011)
Skema Pretreatment
Skema Final Treatment
1.6 Pengolahan
1. Proses pengolahan perlu melakukan tindakan pencegahan untuk mengurangi pencemaran pada seluruh tahap pengolahan termasuk tahap sebelum proses sterilisasi. Pembuatan produk yang berasal dari sumber mikrobiologis hendaklah tidak diproses atau diisi di area yang digunakan untuk pembuatan obat lain namun, vaksin yang mengandung organisme mati atau ekstrak bakterial dapat diisikan ke dalam wadah-wadah, di dalam bangunan dan fasilitas yang sama dengan obat steril lain, setelah proses inaktivasi yang tervalidasi dan pembersihan menurut prosedur yang tervalidasi.
2. Validasi proses aseptis hendaklah mencakup uji simulasi proses menggunakan media pertumbuhan (media fill). Pemilihan media pertumbuhan hendaklah dilakukan berdasarkan bentuk sediaan dan selektivitas, kejernihan, konsentrasi dan cara sterilisasi yang sesuai untuk media tersebut. Validasi proses aseptis dilakukan dalam kondisi produksi normal.Uji simulasi aseptis hendaklah dilakukan semirip mungkin dengan proses aseptis pada produksi rutin dan termasuk semua wadah dan peralatan yang digunakan. Perlu dilakukan pada kombinasi yang diperlukan dari ukuran wadah (ampul, vial, dsb.)termasuk lebar mulut wadah dan kecepatan pengisian (lebih dianjurkan kombinasiekstrim).Bila proses produksi aseptis dimulai pada saat pencampuran bahan sampai denganpengisian, maka proses simulasi hendaklah mencakup seluruh proses, tangki danwadah yang digunakan. Uji simulasi hendaklah menggambarkan semua kondisi pada kasus terburuk (worstcase) yang mungkin terjadi pada produksi normal, misal: pergantian personil, frekuensi istirahat, lampu mati, mesin rusak dan teknisi masuk ke dalam ruang aseptis, dan lain-lain. Volume yang terbesar sering dianggap merupakan kondisi worst case karena mulutwadah produk paling lebar, pengisian paling lambat sehingga produk makin lama terpapar di lingkungan. Tetapi ada beberapa perkecualian dalam hal pengisian
kedalam wadah yang kecil misal ampul 1 ml, pada kasus ini proses pengisianmembutuhkan waktu paling cepat dibandingkan dengan ampul volume lain sehinggaada risiko wadah terguling atau tersendat yang menyebabkan intervensi manual dilakukan lebih sering dari biasanya, disini perlu dilakukan uji simulasi.Volume pengisian hendaklah cukup untuk memungkinkan media membasahi seluruh permukaan wadah saat wadah dibalik dan memungkinkan pendeteksian pertumbuhan mikroba dalam wadah.Bila ukuran bets produksi lebih kecil dari atau sama dengan 3000 unit maka jumlahminimal yang harus diisikan pada uji simulasi adalah sama dengan ukuran bets.Simulasi proses dengan media pertumbuhan untuk validasi awal dan tiap kali terjadi perubahan proses kritis (untuk proses produksi/ pencampuran aseptis), ukuran wadah baru, perubahan shift, penambahan personil, alat baru atau modifikasi alat yang langsung kontak dengan produk, dan atau modifikasi sistem tata udara, hendaklahdilakukan 3 kali untuk tiap shift dan proses.Sedangkan untuk revalidasi dapat dilakukan 1 kali untuk tiap shift dan proses tiap 6 bulan sekali.Bila ada kegagalan atau pertumbuhan pada hasil media pertumbuhan, hendaklah dilakukan identifikasi jenis cemaran dan dibandingkan cemaran yang mungkindiperoleh dari pemantauan lingkungan dan personil.Inkubasi hendaklah dilakukan pada 2 (dua) suhu yaitu:
a. 20°C – 25°C selama 7 hari pertama b. 30°C – 35°C untuk 7 hari berikutnya
Suhu inkubasi lain hendaklah berdasarkan data pendukung yang tervalidasi.Sebelum inkubasi diawali dan saat/ setelah pengamatan pada hari ke 7 wadah dibolakbalik agar larutan media dapat membasahi seluruh permukaan wadah.Pengamatan hendaklah dilakukan pada hari ke 8 (setelah inkubasi pada suhu 20°C –25°C sebelum inkubasi suhu 30°C – 35°C), bila memungkinkan, dan setelah hari ke 14. Target hendaklah dengan pertumbuhan nol tetapi tingkat kontaminasi kurang dari 0,1 % dengan tingkat kepercayaan 95 % yang dapat diterima. Industri farmasi hendaklah menentukan batas waspada dan batas bertindak. Tiap
kontaminasi hendaklah diinvestigasi.Peringatan harus diberikan bahwa dengan melaksanakan validasi tidak berarti dapat melakukan kompromi terhadap proses. Hendaklah dilakukan kontrol negatif dan kontrol positif minimal menggunakan 1 (satu) bakteri dan 1 (satu) kapang. Media pertumbuhan yang dipakai hendaklah lulus Growth Promotion Test (GPT) dengan menggunakan 10 – 100 CFU mikroba gram positif, gram negatif, bakteri anaerob, kapang, dan ragi seperti:
a. Bacillus subtilis atau Clostridium sporogenes; b. Staphylococcus aureus;
c. Pseudomonas aeroginosa; d. Candida albicans;
e. Aspergillus niger.
Pemilihan media hendaklah juga mempertimbangkan kemampuannya menumbuhkan mikroorganisme lingkungan, apabila ada riwayat penemuan kontaminasi lingkungan. Hendaklah dilakukan GPT pada media yang dipakai untuk uji simulasi pada akhir masa inkubasi untuk membuktikan bahwa media akan dapat menumbuhkan mikroba bila ada kontaminasi. Mikroba harus tumbuh dalam waktu 5 hari pada suhu inkubasi yang dipakai.Sediaan tetes mata atau telinga biasanya dikemas dalam wadah plastik. Wadah, penetes, tutup dan overseal (bila dipakai) dicuci dan disterilkan sesuai pada produksirutin. Sebagai pengganti sterilisasi dengan panas, dipakai sterilisasi dengan radiasi atau etilen oksida untuk wadah dan perangkatnya.Wadah plastik yang buram akan menghambat pendeteksian pertumbuhan, dalam hal ini seluruh isi wadah hendaklah dituang ke dalam wadah jernih saat pengamatan.
3. Uji simulasi proses hendaklah dilakukan semirip mungkin dengan proses rutin pembuatan aseptis dan mencakup semua langkah kritis pada tahap pembuatan berikut. Perlu juga dipertimbangkan berbagai intervensi yang diperkirakan akan terjadi saat produksi normal termasuk kasus terburuk. Bila ditemukan pertumbuhan pada uji simulasi proses, perlu dilakukan kajian risikoterhadap mutu produk, terutama pemastian sterilitas (sterility assurance) terhadap bets yang dibuat di antara 2 media fill.
4. Uji simulasi proses sebagai validasi awal hendaklah dilakukan dengan tiga uji simulasi berturut-turut yang berhasil per shift, dan diulangi dengan interval yang ditetapkan dan bila ada perubahan signifikan pada sistem tata udara, peralatan, proses dan jumlah shift. Biasanya uji simulasi proses dilakukan dua kali setahun untuk tiap shift dan proses. 5. Jumlah wadah yang digunakan untuk media fill hendaklah cukup
memungkinkan evaluasi absah. Untuk bets ukuran kecil, jumlah wadah untuk media fill hendaklah minimal sama dengan ukuran bets produk. Target hendaklah dengan pertumbuhan nol dan ketentuan berikut hendaklah diterapkan:
a. Bila mengisi kurang dari 5.000 unit, tidak boleh ditemukan unit tercemar;
b. Bila mengisi 5.000 sampai dengan 10.000 unit:
1) Satu (1) unit tercemar hendaklah diikuti dengan investigasi dan pertimbangan untuk mengulang media fill;
2) Dua (2) unit tercemar merupakan pertimbangan untuk dilakukan validasi ulang setelah investigasi;
3) Bila mengisikan lebih dari 10.000 unit: 4) Satu (1) unit tercemar hendaklah dinvestigasi;
5) Dua (2) unit tercemar merupakan pertimbangan untuk dilakukan validasi ulang setelah investigasi.
6. Pencemaran yang terjadi sesekali pada pengisian dengan jumlah berapapun, mungkin merupakan indikasi pencemaran dalam konsentrasi rendah dan hendaklah dianggap mempunyai dampak pada pemastian sterilitas (sterility assurance) dari bets yang diproduksi setelahmedia fill terakhir yang dinyatakan sukses. Kandungan mikroba awal diperoleh dengan pemeriksaan bioburden yang dilakukanantara lain sebelum proses penyaringan larutan, dan terhadap hasil pemeriksaantersebut dilakukan analisis tren.
7. Perhatian hendaklah diberikan bahwa dengan melaksanakan validasi tidak berarti dapat melakukan kompromi terhadap proses.
8. Untuk menghindarkan penyebaran partikel dan mikroba secara berlebihan, kegiatan dalam area bersih, terutama saat berlangsung proses aseptis, hendaklah dibatasi dan gerakan personil hendaklah terkendali, hati-hati dan sistematis. Suhu dan kelembaban lingkungan hendaklah tidak tinggi sehingga mengganggu kenyamanan akibat sifat pakaian yang dikenakan.
9. Cemaran mikroba bahan awal hendaklah minimal. Spesifikasi bahan awal hendaklah mencakup persyaratan kandungan mikroba bila kebutuhan untuk itu telah ditunjukan melalui hasil pemantauan.
10. Wadah dan bahan yang dapat membentuk partikel hendaklah dibatasi jumlahnya di dalam area bersih dan disingkirkan saat proses aseptis sedang berlangsung.
11. Di mana dapat dilakukan hendaklah diambil tindakan untuk mengurangi kontaminasi partikulat terhadap produk akhir/jadi.
12. Komponen, wadah dan peralatan, setelah proses pembersihan/pencucian akhir, hendaklah ditangani sedemikian rupa sehingga tidak terjadi rekontaminasi.
13. Interval antara pencucian dan pengeringan serta sterilisasi komponen, wadah dan peralatan maupun antara sterilisasi dan penggunaannya hendaklah sesingkat mungkin dan diberi batas waktu yang sesuai dengan kondisi penyimpanan tervalidasi Batas waktu yang sesuai setelah pembersihan, sterilisasi, penyimpanan danpenggunaan komponen, wadah dan peralatan hendaklah ditetapkan melalui validasi. 14. Jarak waktu antara awal pembuatan larutan dan sterilisasi atau filtrasi
melalui filter mikroba hendaklah sesingkat mungkin. Batas waktu maksimum hendaklah ditentukan dengan mempertimbangkan komposisinya dan metode penyimpanan yang ditentukan. Kecuali dilakukan tindakan khusus, volume larutan ruahan hendaklah tidak lebih besar daripada jumlah yang dapat diisi dalam satu hari dan hendaklah diisi ke dalam wadah akhir serta disterilisasi dalam satu hari kerja. Tindakan penyimpanan khusus yaitu dalam wadah yang tertutup rapat dan berada dibawah kondisi udara laminar. Untuk proses aseptis,
hal ini hendaklah dibuktikandengan simulasi menggunakan validasi media fill. Larutan yang tersimpan dan diisikanpada hari yang berbeda hendaklah dipisahkan dengan penandaan lot tersendiri dan uji sterilitas terpisah.
15. Tahap pengolahan komponen, wadah produk ruahan dan peralatan hendaklah diberi identitas yang benar.
16. Semua gas yang dialirkan ke dalam larutan atau digunakan untuk menyelimuti produk hendaklah dilewatkan melalui filter penyaring mikroba. Filter gas hendaklah memakai filter hidrofob untuk menghindarkan pertumbuhanmikroba.
17. Bioburden hendaklah dipantau sebelum proses sterilisasi. Hendaklah ditetapkan batas bioburden segera sebelum proses sterilisasi yang dikaitkan dengan efisiensi metode sterilisasi yang digunakan. Penentuan bioburden hendaklah dilakukan terhadap tiap bets produk, baik yang diproses dengan sterilisasi akhir maupun secara aseptis. Bila parameter sterilisasi overkill ditetapkan untuk produk dengan sterilisasi akhir, pemantauan bioburden boleh hanya secara berkala dengan interval menurut jadwal yang sesuai. Untuk sistem pelulusan parametris, penentuan bioburden hendaklah dilakukan terhadap tiap bets dan dikategorikan sebagai pengujian selama-proses. Bila dipersyaratkan, hendaklah dilakukan pemantauan terhadap cemaran endotoksin. Semua sediaan cair, khususnya larutan infus volume besar, hendaklah dilewatkan melalui filter mikroba yang, jika mungkin, dipasang dekat sebelum proses pengisian. Kontribusi bioburden berbagai bahan awal dan bahan pengemas serta prosespembuatan sebelum sterilisasi hendaklah dipahami dan dikendalikan. Pemantauan danstrategi pengendalian termasuk pemantauan berkala dan trending bioburden sebelumlangkah pengurangan apa pun dari bioburden hendaklah ditetapkan dan dijustifikasimelalui proses analisis risiko. Volume sampel hendaklah dijustifikasi denganmemperhitungkan tingkat kontaminasi yang diperkirakan.Bioburden produk hendaklah
ditentukan paling sedikit sebelum proses sterilisasi akhir.Penetapan kriteria keberterimaan untuk bioburden hendaklah berdasarkan tahapsterilisasi; tingkat pemastian sterilisasi (Sterility Assurance Level/ SAL) 106 harusdicapai. Hasil pemeriksaan bioburden hendaklah menjadi parameter pelulusan produkjadi (kecuali apabila menggunakan siklus overkill untuk sterilisasi akhir).Pengkajian risiko hendaklah dilakukan untuk penetapan kebutuhan studi endotoksin.Apabila diperlukan, endotoksin hendaklah ditentukan juga bagi unit produk yang diisiterakhir.Sterilisasi akhir: Untuk sterilisasi akhir, nilai F0 harus diperhitungkan. Pengambilansampel hendaklah dilakukan terhadap wadah yang sudah terisi sebelum sterilisasi.Untuk proses sterilisasi overkill pada produk dengan sterilisasi akhir, industri hendaklahmenjelaskan interval yang dipilih untuk pengujian bioburden.Proses aseptis : Untuk sterilisasi dengan filtrasi, studi efektifitas penyaring harusdiperhitungkan saat menentukan kriteria keberterimaan bioburden sebelumpenyaringan. Ini berarti jika digunakan dua penyaringan yang berurutan, maka sampelproduk hendaklah diambil sebelum penyaringan tahap akhir, bila dimungkinkan secarateknis, contoh: penyaringan pertama ditampung dalam tangki ruahan, penyaringankedua terjadi segera sebelum pengisian. Namun, jika digunakan sistem dua-tahappenyaringan (penyaring kedua dipakai sebagai pengaman, jika penyaring pertamamengalami kegagalan maka persyaratan SAL tetap dapat tercapai), pengambilansampel hendaklah dilakukan sebelum masing-masing proses penyaringan tanpamengompromikan tahap penyaringan. Industri hendaklah menjelaskan pendekatannyajika pengambilan sampel dilakukan sebelum tahap penyaringan pertama.
18. Bilamana larutan dalam air disimpan dalam tangki tertutup rapat, semua katup pelepas tekanan hendaklah dilindungi misal dengan filter udara mikroba hidrofobik.
19. Semua komponen, wadah, peralatan dan barang lain yang diperlukan dalam area bersih, di mana proses aseptis berlangsung, hendaklah disterilkan dan dimasukkan ke area bersih melalui alat sterilisasi berpintu-ganda yang dipasang menyatu pada dinding, atau melalui suatu prosedur yang dapat mencapai tujuan yang sama yaitu tidak menimbulkan kontaminasi.misalnya pembungkusan tiga lapis (triple wrapping), mungkin dapat diterima.Peralatan dan bahan/ barang lain hendaklah sedapat mungkin disterilkan melaluisterilisator berpintu-ganda yang berhubungan langsung dengan area Kelas A. Bilasterilisator tidak langsung berhubungan dengan lokasi di mana proses aseptisberlangsung, peralatan dan bahan/ barang lain hendaklah selalu secara kontinu dijagadi bawah udara Kelas A selama transfer dari sterilisator sampai dengan penyimpananatau pemakaian. Bisa dipakai kereta (trolley) terlindung dengan aliran udara aktifmaupun pasif. Saat kereta otoklaf atau oven dikeluarkan dari sterilisator ke dalam ruang Kelas B, hendaklah tersedia UDAF zona A di depan pintu sehingga semua itemselalu di bawah udara Kelas A sampai peralatan atau bahan dingin.Bila perlindungan kelas A tidak dapat disediakan untuk komponen atau bahan yang diotoklaf, maka hendaklah dilakukan pembungkusan berlapis, menggunakan bahanpembungkus untuk otoklaf, yang memungkinkan penghilangan udara/ penetrasi uappanas dan penghilangan kondensat di samping dapat mempertahankan sterilitasisinya.Bahan yang disterilkan dengan metode lain misal radiasi sinar Gamma atau etilenoksida hendaklah dilindungi dengan pembungkusan yang tepat untuk mempertahankanintegritas sterilitas di luar lingkungan Kelas A. Bahan ini hendaklah dimasukkan ke areaproses aseptis melalui rongga transfer (misal passbox) dengan sistem interlock padapintu-pintunya untuk menghindarkan biokontaminasi lingkungan Kelas A.Permukaan kemasan dan tangki hendaklah didisinfeksi (misal menggunakan lorong UV, cairan disinfektan, VPHP atau elektron beam) yang tervalidasi untuk
menghindaribiokontaminasi terhadap lingkungan kelas A.Transfer bahan terbungkus ke dalam ruang Kelas A hendaklah dilakukan sedemikianrupa sehingga kemasan luar dapat dibuka tanpa mengontaminasi lingkungan Kelas A pada saat produk, permukaan yang kontak dengan produk, bahan pengemas/ penutup terpapar ke lingkungan. Pada saat transfer, hendaklah dihindarkan terpaparnya bahanyang terbuka bungkusnya ke lingkungan di luar zona Kelas A. 20. Prosedur pengisian secara aseptis hendaklah diverifikasi ulang tiap 6
(enam) bulan sekali melalui media fill atau bila dilakukan perubahan baik pada proses maupun pada peralatan yang sudah tervalidasi. Efikasi dari suatu prosedur baru hendaklah divalidasi. Validasi ini hendaklah diverifikasi pada interval yang dijadwalkan berdasarkan riwayat kinerja atau bila ada perubahan signifikan pada proses atau peralatan (CPOB, 2012).
1.7 Sterilisasi
Sterilisasi adalah suatu proses untuk membuat ruang / benda menjadi steril atau suatu proses untuk membunuh semua jasad renik yang ada, sehingga jika ditumbuhkan di dalam suatu medium tidak ada lagi jasad renik yang dapat berkembang biak. Sterilisasi harus dapat membunuh jasad renik yang paling tahan panas yaitu spora bakteri. Sterilisasi juga merupakan proses penghilangan semua jenis mikroorganisme hidup, dalam hal ini (Protozoa, bakteri, fungi, mycoplasma, virus) yang terdapat pada suatu benda.
Tujuan obat dibuat steril (seperti obat suntik) karena berhubungan langsung dengan darah atau cairan tubuh dan jaringan tubuh yang lain dimana pertahanan terhadap zat asing tidak selengkap yang berada di saluran cerna /gastrointestinal, misalnya hati yang dapat berfungsi untuk menetralisir / menawarkan racun (detoksikasi = detoksifikasi).
Diharapkan dengan steril dapat dihindari adanya infeksi sekunder. Dalam hal ini tidak berlaku relatif steril atau setengah steril , hanya ada dua pilihan yaitu steril dan tidak steril.Sediaan farmasi yang perlu disterilkan
adalah obat suntik / injeksi, tablet implant dan sediaan untuk mata seperti tetes mata /Guttae Ophth., cuci mata / Collyrium dan salep mata / Oculenta, Selain itu tujuan sediaan di sterilisasi adalah untuk mencegah peralatan cepat rusak, mencegah terjadinya infeksi silang, Menjamin kebersihan alat, menetapkan produk akhir dinyatakan sudah steril dan aman digunakan.
Macam-macam sterilisasi : 1. Sterilisasi uap(Cara Panas Basah)
Proses sterilisasi thermal yang menggunakan uap jenuh dibawah tekanan selama 15 menit pada suhu 121o. Kecuali dinyatakan lain, berlangsung di suatu bejana yang disebut otoklaf, dan mungkin merupakan proses sterilisasi paling banyak dilakukan.
Alat : Disebut otoklaf, yaitu suatu panci logam yang kuat dengan tutup yang berat, mempunyai lubang tempat mengeluarkan uap air beserta krannya, termometer, pengatur tekanan udara, klep pengaman.
Sterilisasi cara panas basah (pemanasan dalam otoklaf) hanya sesuai untuk bahan yang terbasahi dengan air dan formula dalam air. Suhu dan tekanan hendaklah digunakan untuk memantau proses sterilisasi. Instrumen pengendali hendaklah independen terhadap instrumen pemantau dan lembar pencatat. Pemakaian sistem pengendali dan pemantau otomatis hendaklah tervalidasi untuk memastikan pencapaian persyaratan proses kritis. Kesalahan pada sistem dan siklus hendaklah terdeteksi dan/atau tercatat oleh sistem dan diamati oleh operator. Pembacaan indikator suhu independen, hendaklah diperiksa secara rutin dan dibandingkan dengan pencatat grafik selama proses sterilisasi. Bila digunakan sterilisator yang dilengkapi dengan drainase pada dasar chamber, perlu juga dilakukan pencatatan suhu pada posisi tersebut selama proses sterilisasi. Bila fase vakum merupakan bagian dari siklus sterilisasi, uji kebocoran pada chamber hendaklah dilakukan secara berkala.
2. Sterilisasi panas kering
Sterilisasi cara ini menggunakan suatu siklus Oven modern yang dilengkapi udara yang dipanaskan dan disaring. Rentang suhu khas yang
dapat diterima di dalam bejana sterilisasi kosong adalah lebih kurang 15o, jika alat sterilisasi beroperasi pada suhu tidak kurang dari 250o .
Alat : Oven yaitu lemari pengering dengan dinding ganda, dilengkapi dengan termometer dan lubang tempat keluar masuknya udara, dipanaskan dari bawah dengan gas atau listrik.
Ciri-ciri pemanasan kering :
a. Yang dipanaskan adalah udara kering
b. Proses pembunuhan mikroba berdasarkan oksidasi O2 udara
c. Suhu yang digunakan lebih tinggi, kira-kira 150o. Satu gram udara pada suhu 100o, jika didinginkan menjadi 99o hanya membebaskan 0,237 kalori.
d. Waktu yang diperlukan lebih lama, antara 1 jam sampai 2 jam, kecuali pemijaran.
e. Digunakan untuk sterilisasi bahan obat / alat yang tahan pemanasan tinggi.
3. Sterilisasi gas
Bahan aktif yang digunakan adalah gas etilen oksida yang dinetralkan dengan gas inert, tetapi keburukan gas etilen oksida ini adalah sangat mudah terbakar, bersifat mutagenik, kemungkinan meninggalkan residu toksik di dalam bahan yang disterilkan, terutama yang mengandung ion klorida.
Pemilihan untuk menggunakan sterilisasi gas ini sebagai alternatif dari sterilisasi termal, jika bahan yang akan disterilkan tidak tahan terhadap suhu tinggi pada sterilisasi uap atau panas kering.
Proses sterilisasinya berlangsung di dalam bejana bertekanan yang didesain seperti pada otoklaf dengan modifikasi tertentu. Salah satu keterbatasan utama dari proses sterilisasi dengan gas etilen oksida adalah terbatasnya kemampuan gas tersebut untuk berdifusi sampai ke daerah yang paling dalam dari produk yang disterilkan.
Metode sterilisasi ini hendaklah hanya digunakan bila cara lain tidak dapat diterapkan. Selama proses validasi hendaklah dibuktikan bahwa tidak ada akibat yang merusak produk. Kondisi dan waktu yang
diberikan untuk menghilangkan gas hendaklah ditentukan untuk mengurangi gas residu dan zat hasil reaksi sampai pada batas yang dapat diterima yang sudah ditetapkan untuk tiap produk atau bahan.
Berbagai gas dan fumigan dapat digunakan untuk sterilisasi (misal etilen oksida, uap hidrogen peroksida). Etilen oksida hendaklah digunakan hanya bila tidak ada metode lain yang dapat dipakai. Kontak langsung antara gas dan sel mikroba adalah esensial; tindakan pencegahan hendaklah dilakukan untuk menghindarkan organisme yang mungkin terperangkap dalam bahan misal dalam kristal atau protein yang dikeringkan. Jumlah dan sifat bahan pengemas dapat memengaruhi proses secara signifikan.
4. Sterilisasi dengan radiasi ion
Sterilisasi dengan cara radiasi terutama digunakan untuk bahan dan produk yang peka terhadap panas. Banyak obat dan bahan pengemas peka terhadap radiasi, sehingga metode ini hanya dipakai jika terbukti tidak berdampak merusak yang dibuktikan melalui eksperimen. Biasanya radiasi ultraviolet tidak diterima sebagai metode sterilisasi. Jika sterilisasi cara radiasi dilakukan oleh pihak luar, maka industri bertanggung jawab atas pemenuhan persyaratan yang tercantum pada Butir dan proses sterilisasi tervalidasi. Hendaklah ditetapkan tanggung jawab dari perusahaan yang melakukan radiasi (misal penggunaan dosis yang benar).
Cara ini dilakukan jika bahan yang disterilkan tidak tahan terhadap sterilisasi panas dan khawatir tentang keamanan etilen oksida.Keunggulan sterilisasi ini adalah reaktivitas kimia rendah, residu rendah yang dapat diukur serta variabel yang dikendalikan lebih sedikit. 5. Sterilisasi dengan cara filtrasi
Sterilisasi larutan yang labil terhadap panas sering dilakukan dengan penyaringan menggunakan bahan yang dapat menahan mikroba, hingga mikroba yang dikandungnya dapat dipisahkan secara fisika.
Perangkat penyaring umumnya terdiri dari suatu matriks berpori bertutup kedap atau dirangkaikan pada wadah yang tidak permeable.Efektivitas penyaring media atau penyaring subtrat tergantung pada ukuran pori matriks, daya adsorpsi bakteri dari matriks dan mekanisme pengayakan.
Larutan disaring melalui penyaring bakteri steril, diisikan ke dalam wadah steril, kemudian ditutup kedap menurut teknik aseptik.
1.8 Filtrasi untuk Bahan yang Tidak Dapat Disterilkan dalam Wadah Akhirnya
Filtrasi merupakan metode sterilisasi yang digunakan dalam produksi steril secara aseotis. Tetapi filtrasi dianggap tidak cukup untuk memenuhi sterlititas apabila suatu produk dapat dilakukan sterilisasi pada wadah akhirnya. Untuk produk yang tidak dapat dilakukan sterilisasi pada wadah akhirnya maka filtrasinya menggunakan cara khusus seperti yang telah disebut dalam CPOB (2012) yaitu :
1. Untuk produk berupa larutan/cairan difiltrasi ke dalam wadah yang telah lebih dahulu disterilkan. Filtrasi dilakukan menggunakan filter dengan ukuran <0,22 mikro meter. Dapat dilakukan pemanasan sebagai pelengkap proses filtrasi
2. Dilakukan filtrasi kembali karena beranggapan bahwa dengan sekali filtrasi saja masih memiliki resiko kontaminasi. Filtrasi ini digunakan pula sebagai pelengkap filtrasi awal
Filtrasi kedua dilakukan menggunakan filter yang steril. Selain itu, untuk menurunkan risiko kontaminasi terhadap produk maka filtrasi kedua dilakukan sedekat mungkin ke titik pengisiannya. Filter yang digunakan sebaiknya filter yang tidak mempengaruhi produk, dimana filter tidak meninggalkan serat atau bahan filter ke pada produk.
Dalam memastikan apakah suatu filter masih layak digunakan atau tidak dalam filtrasi, maka diperlukan suatu uji yaitu uji integritas filter. Terdapat beberapa uji integeritas filter, antara lain:
1. Uji Bubble Point
Uji bubble point adalah uji yang dirancang untuk menentukan tekanan di mana sebuahaliran kontinu gelembung yang awalnya terlihat di hilir
(downstream) filter yang terbasahi dalam suatutekanan gas. Untuk melakukan uji bubble point, gas diterapkan ke satu sisi darifilter yang telah terbasahi, dengan tabung downstream filter yang terendam di ember air. Filter harus dibasahi sehingga air mengisi semua rongga di dalam media filter. Ketika tekanan gas diterapkan ke satu sisi membran,gas akan larut ke dalam air. Downstream dari filter memiliki tekanan yang lebih rendah. Oleh karena itugas di dalam air di sisi downstream didorong keluar dari solusi. Ketika tekanan gas hulu (upstream) meningkat, aliran difusi downstream juga meningkat secara proporsional. Di beberapa titik, tekanan menjadi cukup besar untuk mengusir air dari satu atau lebih pori dan membangun jalan untuk aliran udara.Akibatnya, aliran gelembung terlihat keluar dari tabung yang terendam di ember air. Tekanan pada saat aliran gelembung tersebut terlihat disebut sebagaibubble point.
2. Uji Diffusive Flow
Ketika tekanan udara atau nitrogen diterapkan ke satu sisi dari filter yang dibasahi,molekul gas di sisi hulu (upstream) yang bertekanan tinggi akan larut dalam lapisan air dalampori sesuai dengan Hukum Henry. Gas yang keluar dari solusi di sisi hilir (downstream) sebagai gelembung mikro (micro bubbles) atau perpindahan volume,lagi-lagi menurut Hukum Henry, yang mana gas akan mengatur tingkatpembentukan microbubble atau perpindahan cairan, yaitu secara eksperimental. Dengan demikian,tingkatdiffusive airflow atau aliran udara difusif adalah fungsi dariperbedaan tekanan transmembran .Percobaan telah menunjukkan bahwa tingkat aliran udara difusi dengan perbedaan tekanan diferensial berkorelasidengan tingkat retensi organisme tertentu.Pengujian aliran udara difusi dapat dilakukan menggunakan dua cara pengukuran, yaitu single point measurement dan multipoint diffusion measurement.
3. Uji Pressure Hold
Uji pressure hold adalah tes integritas nondestruktif berdasarkan aliran difusi (aliran maju) dari cartridge. Menggunakan pengukur yang sangatakurat , perubahan tekanan hulu (upstream)selama difusi gas
melalui filter terpantau dari waktu ke waktu.Karena yang diukur adalah upstream, resiko sterilitas hilir (downstream) dihilangkan . Nilai ini sangattergantung pada volume sistem sebagaimana hilangnya tekanan akan terjadi karena aliran yang berdifusi melalui cartridge.Dengan mengetahui aliran difusi maksimum cartridge dan volume sistem maka memungkinkan untuk menghitung penurunan maksimum tekanan yang diizinkan.
(Jornitz, 2004) 1.9 Contoh produksi produk steril
1. Sediaan Padat
a. Proses Pembuatan Produk Steril Injeksi Kering
Alur proses produksi sediaan injeksi kering dan serbuk untuk obat suntik dilakukan dengan tahapan :
1) Menyiapkan material equipment yg telah melalui proses aseptis untuk masing-masing bahan aktif dan bahan tambahan.
2) Melakukan pemeriksaan fisiko kimia dan pirogen bahan yang digunakan meliputi pemeriksaan gelas pada ampul, atau vial dan pemeriksaan fisiko kimia pada karet dan plastik.
3) Setelah itu dilakukan pencampuran bahan aktif obat dan eksipien menggunakan homogenizer
4) Setelah itu dilakukan sterilisasi hasil produk parenteral dengan memilih metode sterilisasi yang sesuai
5) Kemudian dilakukan uji produk parenteral dan dilakukan validasi. (Lukas, 2011)
b. Proses Produksi Injeksi Kering yang Terpisah dengan Pelarutnya 1) Dilakukan penghalusan partikel obat dengan menggunakan bola
penggiling atau peralatan lain yang sesuai
2) Selanjutnya dilakukan sterilisasi pada serbuk injeksi dan pelarut dengan metode yang sesuai
3) Dilakukan penempatan serbuk injeksi pada wadah yang cukup besar untuk memungkinkan pengocokan apabila telah direkonstitusi dengan pelarutnya
4) Dilakukan proses akhir yakni pemberian etiket dan penyimpanan dalam tempat yang sesuai.
5) Dilakukan penjagaan suhu penyimpanan sehingga sediaan tetap stabil selama penyimpanan dan sebelum digunakan (lukas, 2011) c. Proses pembuatan injeksi serbuk kering
Untuk sediaan parenteral berbentuk serbuk, serbuk biasanya dibuat secara kristalisasi dan semprot kering (spray drying) aseptik untuk menjamin sterilitas dan bioburden dari bahan secara maksimal. Pada teknik kristalisasi, obat dilarutkan dalam pelarut yang sesuai dan disterilkan dengan cara filtrasi. Dalam kondisi terkendali ditambahkan pelarut steril yang lain dimana obattidak larut. Penambahan pelarut tersebut bertujuan menginduksi proses kristalisasi dari obat. Selanjutnya kristal dipisahkan, lalu dicuci dan dikeringkan. Kemudian diuji dstribusi ukuran partikelnya, kecepatan disolusinya dan identifikasi bentuk kristal yang sesuai sebelum diisikankedalam vial. Jika serbuk obat dibuat secara semprot kering, maka terlebih dahulu dibuat larutan steril dari obat dengan cara yang sama seperti pada kristalisasi secara aseptis. Larutan steril atau lumpuran disemprotkan melalui pengatomisasi (atomizer) dengan diameter lubang kecil kedalam ruangan pengering (steril).Pada saat berkontak dengan aliran udara steril, pelarut secara cepat menguap, dan serbuk yang dihasilkan ditampung dalam suatu ruangan steril.Untuk tujuan ini dapat digunakan bermacam alat teknologi semprot kering.
Untuk bahan aktif obat dan formulasi sediaan yang tidak stabil, apalagi setelah mengalami beberapa proses, maka dapat dilakukan proses liofilisasi (freeze drying), dimana larutan membentuk kue (cake) serbuk yang siap direkonstitusi pada saat akan digunakan (umumnya dengan pelarut air) (Agus, 2009)
d. Liofilisasi Sediaan Parenteral
Proses liofilisasi memiliki tahapan berikut :
1) Melarutkan obat dengan eksipien dalam pelarut yang sesuai, pada umumnya air untuk injeksi.
2) Mensterilkan larutan ruahan dengan cara penyaringan melalui penyaring bakteri ukuran 0,22 µm
3) Pengisian kedalam kontener (ampul, vial) steril secara individual dan menutup kontener dibawah kondisi aseptik secara parsial. 4) Memindahkan kontener tertutup parsial ke liofilizer dan
menempatkannya dalam ruangan pada kondisi aseptic
5) Membekukan larutan dengan cara meletakkan kontener yang ditutup parsial pada rak dingin dalam ruangan pengering beku (freeze drying) atau melakukan prapembekuan (prefreezing) dalam ruangan lain.
6) Aplikasi vakum pada ruangan dan memanaskan rak untuk penguapan air dari keadaan beku
7) Penutupan sempurna vial, biasanya dilakukan secara hidrofilik atau mekanisme pemutaran atau penekanan yang ditempatkan di liofilizer. (Agus, 2009)
2. Sediaan Semipadat
Salep mata atau oculenta adalah gel dengan perubahan bentuk plastis, yang ditentukan untuk digunakan pada mata. Persyaratannya harus steril, tanpa kontaminan mikroba (0 koloni), tidak iritatif serta memiliki daya lekat dan daya sebar baik dan lembut pada mata. Salep mata bersifat hidrofil, mampu beremulsifikasi dengan cairan air mata sehingga distribusinya baik dalam konjungtiva (Voight, 1995)
Pembuatan salep mata harus steril serta berisi zat antimicrobial preservative, antioksidan dan stabilizer. Menurut USP edisi XXV, salep berisi chlorobutanol sebagai antimicrobial dan perlu bebas bahan partikel yang dapat membahayakan jaringan mata. Sebaliknya, dari EP (2001) DAN bp (2001) ada batasan ukuran partikel, yaitu setiap 10 mikrogram zat aktif tidak boleh mempunyai partikel >90 nm, tidak boleh lebih dari dua partikel >50nm, dan tidak boleh lebih dari 20,25 nm (Lukas, 2011).
Dalam memformulasikan sediaan untuk mata, baik secara industry maupun extempore perlu diperhatikan sejumlah factor seperti tipe sediaan dan cara penggunaannya, aktivitas dan stabilitas bahan aktif obat, pengaturan tonisitas, pilihan metode sterilisasi, dan pengemasan untuk sediaan obat mata yang dibuat (Agoes, 2009)
Sterilitas adalah salah satu persyaratan penting untuk larutan optalmik. Karena stabilitas terjadap panas dari sediaan mata-extemporer sering tidak diketahui, cara pembuatan obat mata ini sebaiknya disterilkan dengan cara filtrasi melalui penyaringan bakteri. kerugian cara sterilisasi ini adalah tidak mampu menghilangkan kontainasi virus. Cara ini juga sangat bergantung pada operasional teknik aseptic yang dilakukan dalam sterilisasi larutan dan pengemasannya.
Salep mata adalah salep yang digunakan pada mata. Pada pembuatan salep mata harus diberikan perhatian khusus. Sediaan dibuat dari bahan yang sudah disterilkan dengan perlakuan aseptic yang ketat serta memenuhi syarat uji sterilitas. Bila bahan tertentu yang dgunakan dalam formulasi salep mata tidak dapat disterilkan dengan cara biasa, maka dapat digunakan bahan yang mmenuhi syarat uji sterlitas dengan pembuatan secara aseptic. Salep mata mengandung bahan atau campuran bahan yang sesuai untuk mencegah pertumbuhan atau memusnahkan mikroba yang mungkin masuk secara tidak sengaja bila wadah dibuka pada waktu aplikasi penggunaan, kecuali dinyatakan lain dalam monografi, atau formulanya sendiri sudah bersifat bakteriostatik.
Pembuatan salep mata harus berlangsung pada kondisi aseptik dalam boks laminar untuk menjamin kemurnian mikrobiologis yang disyaratkan. Hal ini mensyaratkan bahwa basis salep yang digunakan pun sedapat mungkin dapat disterilkan. Disarankan untuk menggunakan Vaseline yang mengandung kolesterol yang dapat disterilkan menggunakan udara panas tanpa mengurangi kualitasnya. Juga dimungkinkan dengan menggunakan penyaringan bebas kuman dari basis salep di dalam alat penyaring tekan yang dapat dipanaskan (Voight, 1995) Dasar salep mata yang dipilih tidak boleh mengiritasi mata, memungkinkan difusi obat dalam cairan mata, dan tetap dapat mempertahankan aktivitas obat dalam jangka waktu tertentu pada kondisi penyimpanan yang tepat (usia guna). Vaseline merupakan dasar salep mata yang banyak digunakan. Beberapa bahan dasar salep yang dapat menyerap air mata, bahan dasar yang mudah dicuci dengan air, dan
bahan dasar larut air, dapat digunakan untuk obat yang larut dalam air. Bahan dasar salep seperti ini memungkinkan dispersi obat larut secara lebih baik, tetapi tidak boleh menyebabkan iritasi pada mata (Agoes, 2009).
Untuk menjamin pelepasan bahan obat yang baik, disrankan untuk membuat salep suspensi. Dalam hal ini ukuran partikel bahan obat sangat diperhatikan untuk mencegah iritasi terhadap mata dan meningkatkan efektifitas kerjanya, digunakan serbuk yang dimikronisasikan atau serbuk dengan karakteristik ukuran butir yang sama. Di dalam industry bisa menggunakan mesin penggiling kemudian dilanjutkan dengan proses fraksionasi partikel.
Sedangkan pembuatan salep mata dengan formulasi emulsi atau bentuk larutan dalam air mensyaratkan kelarutan bahan obat dalam air sangat baik sehingga mudah dihablurkan (Voight, 1995).
Bahan tambahan yang ditambahkan ke dalam dasar salep mata berbentuk larutan atau serbuk halus. Salep mata harus bebas dari partikel kasar dan harus memenuhi syarat kebocoran dan partikel logam pada uji salep mata. Wadah container untuk salep mata harus dalam keadaan steril pada waktu pengisian dan penutupan serta harus tertutup rapat dan disegel untuk menjamin sterilitas pada penggunaan pertama obat (Agoes, 2009).
Pengemasan yang paling baik untuk salep mata adalah tube, dengan pertimbangan kontaminasi saat pemakaian lebihkecil dibandingkan wadah salep pada umumnya karena isi salep langsung terpapar lingkungan luar, sedangkan pada tube tidak. Selain itu terlindung dari cahaya. Penggunaan bahan tube harus diperhatikan terutama agar tidak mempengaruhi atau bereaksi dengan salep, umumnya digunakan tube dari bahan nonlogam. Salep mata mempunyai waktu penyimpanan yang sempit karena bergantung pada stabilitas bahan kimia bahan obat dan kemungkinan terjadinya perubahan ukuran partikel atau rekristalisasi sehingga perlu dilakukan pengujian ukuran partikel secara berkala
selama proses penyimpanan. Namun, lebih disarankan salep mata segera didistribusikan setelah diproduksi (Voight, 1995)
3. Sediaan cair
a. Injeksi dan Infus
Apabila formula suatu produk parenteral (injeksi atau infus) telah ditentukan, meliputi pelarut atau pembawa yang tepat, maka sejak awal proses pembuatan sediaan harus mengikuti prosedur aseptik. Proses aseptik dilakukan karean produk parenteral yang akan digunakan harus bebas dari mikroorganisme, mulai dari pelarut (air) dan bahan-bahan zat aktif hingga bahan tambahan (material equipment).
Setelah memproses bahan tambahan, kita memerlukan pemeriksaan pendahuluan fisika-kimia dan pirogen masing-masing bahan yang digunakan. Sebaliknya, dilakukan pemeriksaan gelas pada ampul atau vial dan pemeriksaan fisika kimia pada karet atau plastik.
Setelah mencampur beberapa zat aktif dengan bahan tambahan menjadi bentuk larutan, kemudian dilakukan penyaringan sampai jernih berkilauan dengan sintered glass, porselen, kertas saring yang tebal, atau saringan jenis membran. Sesudah penyaringan, pindahkan larutan secepat mungkin dan sesedikit mungkin terjadi pemaparan mikroba dan partikel ke dalam wadah akhir, lalu tutup dengan rapat. Selanjutnya, hasil produk parenteral disterilkan menggunakan metode sterilisasi yang sesuai. Tahapan akhir yaitu diberi etiket pada produk akhir, dan dilakukan uji produk parenteral serta validasi.
Pembuatan sediaan suspensi injeksi atau infus dengan cara menghaluskan obat hingga menjadi serbuk yang sangat halus menggunakan bola penggiling atau peralatan lain yang sesuai. Selanjutnya, serbuk halus disuspensikan dalam cairan yang tidak melarutkan zat aktif. Seringkali membutuhkan pensterilan masing-masing komponen suspensi secara terpisah sebelum mencampurkannya karena terkadang keutuhan suspensi dirusak oleh pensterilan dengan autoklaf. Pensterilan suspensi parenteral dengan autoklaf dapat
mengubah viskositas produk. Perubahan viskositas akan mempengaruhi kemampuan pembawa sebagai pensuspensi atau mengubah ukuran partikel zat yang disuspensikan, yang berarti mengubah terapi sediaan. Jika suspensi tetap tidak berubah oleh autoklaf, maka kita dapat menggunakannya untuk mensterilkan produk steril.
Pembuatan sediaan emulsi injeksi atau infus dengan cara mencampurkan cairan dengan cairan lain menggunakan alat homogenizer. Alat dapat menghomogenkan sistem dispersi dari emulsi atau suspensi, baik zat padat maupun cair dengan volume yang dapat diproses berkisar dari 0,5 ml sampai lebih dari 25 liter. Selanjutnya, sediaan emulsi disterilkan dengan autoklaf. Umumnya sediaan emulsi rusak bila disterilkan dengan autoklaf sehingga kita harus mensterilkan dengan cara lain.
b. Obat Tetes Mata, Hidung, dan Telinga
Pada produksi sediaan oftalmik, sebaiknya disterilkan dengan cara filtrasi melalui penyaring bakteri. Sterilisasi larutan oftalmik dengan cara penyaringan dapat menjernihkan larutan dengan menghilangkan partikel partikulat dari larutan. Beberapa larutan oftalmik dapat disterilkan dengan autoklaf dalam kontener terakhir, dengan catatan bahwa obat stabil terhadap panas. Apabila digunakan kemasan plastik yang tidak stabil terhadap panas autoklaf, maka kemasan plastik dapat disterilkan dahulu secara sterilisasi radiasi, dan kemasan steril diisi dengan larutan yang sudah disterilkan melalui teknik aseptik (penyaringan). Metode filtrasi adalah cara sterilisasi pilihan yang sering diaplikasikan.
Suspensi Oftalmik
Pada sediaan suspensi oftalmik digunakan bentuk halus (microfine) partikel dengan rentang ukuran sekitar 10 µm dan disuspensikan dalam pembawa air. Sebelumnya, bahan aktif dan bahan tambahan telah disterilkan dengan cara sterilisasi yang sesuai. Selanjutnya, suspensi dibuat dengan cara aseptik sebelum diisikan ke dalam kontener akhir yang sudah disterilkan.
Selanjutnya, pembuatan sediaan otik dan nasal didasarkan pada pembuatan sediaan steril sehingga cara sterilisasi dan teknik aseptik yang digunakan sama dengan cara sterilisasi dan teknik aseptik untuk preparasi obat steril, seperti injeksi.
4. Sediaan Khusus a. Hormon
Hormon merupakan protein aktif yang secara normal diproduksi sendiri oleh tubuh, yang selanjutnya disebut hormone endogen. Namun, pada kondisi patologis tertentu hormon kurang atau tidak diproduksi sehingga memerlukan hormone dari luar (hormon eksogen). Hormon memiliki sifat seperti halnya protein pada umumnya, terutama yang harus diperhatikan dalam industri farmasi hormone adalah struktur hormone sendiri yang tidak stabil terhadap beberapa factor, seperti pH, radiasi, suhu, medium pelarut organik, surfaktan, dan lain-lain.
Stabilitas polipeptida dan protein diklasifikasikan menjadi stabilitas fisika dan kimia, dan selalu memiliki hubungan signifikan antara stabilitas konformasional dan integritas kimia dari setiap molekul. Ketidakstabilan kimia disebabkan oleh pembentukan atau dekstruksi ikatan kovalen dalam molekul polipeptida atau protein. Perubahan ini akan mengganggu struktur primer dari protein dan dapat berdampak pada tingkat struktur lebih tinggi. Sedangkan ketidakstabilan fisika menyebabkan perubahan konformasional (perubahan struktur sekunder dan tersier) sebagai hasil ekspose gangguan structural atau karena stres lingkungan, seperti perubahan suhu dan pH.
Salah satu contoh produk hormon adalah insulin yang diberikan secara injeksi. Injeksi insulin adalah injeksi larutan air insulin yang steril. Injeksi insulin mengandung 100 atau 500 unit insulin setiap mL. label harus menyatakan potensi dalam unti USP per mL dan tanggal daluwarsa (tidak lebih dari 24 bulan setelah dibuat). Sebagai
upaya pengamanan dari penggunaan kadar obat yang tidak benar, pada kemasan dicantumkan kode warna unik untuk tiap-tiap kadar. Sebagai contoh, kode warna untuk semua insulin dari bermacam industry yang mengandung 100 unit/mL berwarna oranye, sedangkan sediaan dengan 500 unit/mL berwarna coklat dengan garis diagonal berwarna putih. Penyimpanan insulin stabil pada kondisi dingin (>15 ºC). pembekuan (freezing) dihindari karena akan menurunkan potensi insulin.
Terdapat dua betuk suspensi injeksi insulin dalam pembuatannya, yaitu insulin NPH (Neutral Protamin Hagedorn) dan ultralente, sedangkan dua pendekatan dalam pembuatan suspensi insulin, sebagai berikut :
1) Kristal berasal dari larutan yang mengandung semua komponen formulasi pada konsentrasi yang sesuai untuk pembuatan forulasi akhir.
2) Tipe proses pembuatan suspensi ultralenta dengan membat suspensi konsentrat Kristal yang kemudian diencerkan dengan pembawa suspensi air yang sesuai untuk menghasilkan suspensi akhir
b. Vaksin
Vaksin merupakan salah satu produk farmasi yang memiliki manfaat yang sangat besar bagi manusia, terutama dalam pencegahan dan pengobatan terhadap berbagai penyakit mematikan yang ditimbulkan oleh virus dan bakteri. Berbagai penyakit mematikan seperti campak, polio, difteri dan tetanus pada manusia telah berhasil diatasi dengan adanya vaksin yang mampu meningkatkan antibodi menjadi lebih kuat dan tahan terhadap penyakit-penyakit tersebut. Keberadaan vaksin mutlak dibutuhkan guna menunjang kesehatan umat manusia.
PT. Bio Farma sebagai salah satu produsen vaksin di dunia serta satu-satunya di Indonesia. Beberapa produk vaksin yang diproduksi oleh PT. Biofarma yaitu vaksin difteri dan tetanus.
(Priambodo, 2012) Beberapa jenis vaksin menurut teknologi pembuatannya antara lain vaksin hidup (live attenuated vaccine), vaksin inaktif (killed vaccine),
vaksin kombinasi, formulasi vaksin baru, vaksin subunit, vaksin rekombinan dan vaksin nukleotida.
1) Vaksin hidup (live attenuated vaccine) merupakan vaksin yang dihasilkan dengan cara melemahkan virus dan mengadaptasi pertumbuhan pada suhu tertentu (33˚C atau 35˚C). Contoh vaksi polio oral Sabin dan vaksin campak (Schwarz)
2) Vaksin inaktif (killed vaccine) merupakan vaksin yang dihasilkan dengan menginaktifkan virus dalam larutan formalin (0,2 % formalin selama 1 jam pada suhu 37˚C. misalnya vaksin polio Salk dan vaksin campak Edmonston
3) Vaksin kombinasi biasanya berisi lebih dari dua jenis antigen. Misalnya vaksin Diphteria, Pertusis dan Tetanus (DPT). Vaksin ini dibuat dengan tujuan mengurangi banyaknya suntikan yang diberikan. 4) Formulasi vaksin baru merupakan vaksin yang dibuat dengan
meningkatkan dosisnya sehingga dapat diberikan dengan satu kali suntikan saja
5) Vaksin subunit adalah perkembangan dari vaksin inaktif dimana mengandung beberapa epitop dari suatu antigen. Dihasilkan dengan cara membuat peptida sintetik yang mirip dengan komposisi antigen tersebut. Contohnya vaksin subunit SPf 66 terhadap malaria
6) Vaksin rekombinan merupakan vaksin yang menggunakan virus sebagai vector. Dibuat dengan cara menyisipkan gen yang mengkode epitop tertentu pada plasmid, kemudian ditransfeksikan ke dalam suatu virus, sehingga terjadi suatu virus rekombinan. Virus rekombinan ini dipakai sebagai vektor gen yang mengekspresikan epitop tertentu dari suatu antigen tadi pada sel mamalia
7) Vaksin polionukleotida merupakan suatu bentuk rekombinan, komposisi antara plasmid dengan genom virus yang sangat konserv (tidak berubah)
(Yuwono, 1995) Sedangkan beberapa klasifikasi produk vaksin menurut Josefsberg (2012) dalam jurnal Vaccine Process Technology sebagai berikut:
Live attenuated virus Smallpox, polio, measles, mumps, rubella, chicken pox, rotavirus, shingles, influenza, and yellow fever
Inactivated purified virus Inactivated polio, japanese encephalitis, hepatitis A, Influenza (seasonal and pandemic), and rabies
Live attenuated bacterium Tuberculosis and typhoid Whole inactivated bacterium Whole cell pertussis Purified protein Acellular pertussis
Purified protein toxoid Tetanus, anthrax, and diphtheria Purified virus-like particles (VLPs) Hepatitis B and human papillomavirus Purified polysaccharide Pneumococcal for adults and typhoid Polysaccharide conjugated to carrier
protein
Pneumococcal for infants, haemophilus type B, and bacterial meningitis
Plasmid DNA Dalam pengembangan
Adenovirus DNA delivery Dalam pengembangan
Dalam CPOB (2012) disebutkan bahwa untuk produksi vaksin BCG dilakukan oleh personil yang sehat dan tidak mengidap infeksi tuberculosis. Apabila terdapat personil yang mengidap tuberculosis maka tidak diperkenankan bekerja di daerah produksi dan seluruh vaksin yang dibuat selama pekerja tersebut bekerja harus dimusnahkan. Selanjutnya dilakukan pemeriksaan terhadap pekerja lainnya untuk mengetahui kemungkinan penularan terinfeksi tuberculosis.
Proses produksi vaksin dilakukan dalam daerah terpisah, tertutup dan menggunakan peralatan tersendiri. Untuk mencegah terjadinya pencemaran yang berasal dari lingkungan maka dalam area produksi steril sebaiknya disiapkan ruang antara yang dirancang khusus untuk menghindari kontaminasi. Selain itu diperlukan ruang kelas atau kelas 100, aliran udara laminar dan efisiensi udara akhir 99,995 %.
Dalam mempelajari literatur tentang produksi vaksin, diketahui bahwa setiap vaksin memiliki perbedaan dalam hal produksinya, tidak ada prosedur khusus untuk pembuatan seluruh vaksin melainkan terkait pada prosedur umum produksi vaksin. Produksi vaksin secara umum, antara lain:
1) Budidaya sel bakteri atau jaringan, pada suhu sekitar 37 ° C di media yang terkadang kompleks
2) Volume kultivasi (budidaya) yang kecil, jika dibandingkan dengan industri yaitu 25-1000 liter.
3) Harus asepsis, serta perlindungan dari operator terhadap infeksi mikroba di bawah budidaya. Pertimbangan pertama dilakukan karena sebagian besar organisme yang terlibat dibudidayakan di media di mana mereka mudah dikontaminasi oleh bakteri; kedua karena strain laboratorium yang digunakan sering dianggap masih menjadi patogen bagi manusia. 4) Produksi zat yang bukan merupakan produk metabolisme utama seperti
etanol dari ragi atau asam sitrat dari Aspergillus niger sehingga perlu dipisahkan (purifkasi).
5) Didapatkannya dari produk akhir (vaksin) yang memaksimalkan keselamatan bagi manusia dimana vaksin sebagai pengobatan preventif.
(Hemert,1971) Kebanyakan vaksin diproduksi dari sel-sel utama yang diambil dari organ dan jaringan hewan, contohnya yaitu sel vero, yang mana merupakan gari sel mamalia pertama yang diperoleh dari monyet hijau Afrika pada tahun 1962. Beberapa contoh vaksin yang diproduksi dari sel ini yaitu vaksin cacar ACAM2000 dan vaksin virus H5N1 (Josefsber, 2012).
Selain itu pembuatan vaksin polio juga menggunakan sel vero sebagai sel utama atau sel pembiakkan, dimana yaitu dengan beberapa tahapan antara lain penyiapan media (sel vero) untuk pengembangbiakkan virus, penanaman/inokulasi virus, pemanenan virus, pemurnian virus, inaktivasi/atenuasi virus. Penyiapan media (sel vero) dilakukan dengan menggunakan mikrokarier yaitu bahan pembawa yang akan mengikat sel tersebut, bahan tersebut adalah NN Diethyl Amino Ethyl (DEAE) dan pada proses selamjutnya sel vero ini harus dilepaskan dari mikrokarier dengan menggunakan enzim tripsin (pankreas babi) selanjutnya pembuangan nutrisi dengan cara dicuci dengan menggunakan larutan PBS buffer. Larutan ini kemudian dinetralkan dengan serum anak sapi (calf serum). Sel – sel vero yang sudah dimurnikan dan dinetralisasi itu kemudian ditambahkan mikrokarier yang baru dan ditempatkan di bioreaktor yang lebih besar dan didalamnya ditambahkan nutrisi dan virus siap untuk dibiakkan. Sel vero yang sudah berkambang biak dan bertambah jumlahnya kemudian dilepaskan lagi dari mikrokriernya dengan
tripsin babi dan proses ini dilakukan berulang – ulang sampai dihasilkan jumlah yang di inginkan.
1.10 Indikator Biologi dan Kimia
Suatu indikator yang digunakan dalam produksi steril berguna untuk menunjukkan adanya kegagalan dari proses sterilisasi. Indikator dalam produksi steril tidak dapat menunjukkan keberhasilan sterilisasi secara sempurna. Beberapa indicator yang digunakan dalam produksi steril antara lain indikator biologis dan kimiawi.
1. Indikator Biologis 2. Indikator Kimiawi
1.11Penyelesaian Produk Steril
Penyelesain produk steril (sterile product finishing) memiliki kekhususan dimana untuk produk beku kering (liofiilisasi) dalam vial proses finishing-nya dilakukan atau ditangani dalam lingkungan kelas A dan secara aseptis.
Penutupan vial untuk produk-produk steril yang menggunakan wadah vial yaitu meliputi pemasangan stopper, pencengkraman alumunium atau crimping dan selajutnya capping. menurut CPOB (2012) dalam proses capping produk steril dapat dilakukan intervensi oleh manusia, hanya saja dengan hal tersebut akan meningkatkan terjadinya resiko kontaminasi. Penggunaan Restricted Access Barrier (RAB) atau isolator dapat digunakan untuk meminimalkan kontaminasi saat dilakukan intervensi oleh manusia.
Uji integritas 100 % atau uji kebocoran dapat dilakukan terhadap produk steril berwadahkan fusi (ampul plastik atau kaca). Uji ini bertujuan untuk memastikan kualitas dan sterilitas dari suatu produk steril. Selain itu dapat pula dilakukan inspeksi fisik (indikator fisik) untuk melihat kontaminasi, benda asing ataupun cacat fisik pada suatu produk steril. Inspeksi dilakukan oleh operator dengan prosedur yang sudah ditetapkan. 1.11Pengawasan Mutu Produk Steril
Dalam mengawasi mutu dari suatu produk steril dapat dilakukan pengujian yaitu uji sterilitas. Uji sterilitas dalam produksi steril di suatu industry dilakukan dengan mengambil sampel produk steril. Sampel yang digunakan yaitu sampel yang mewakili keseluruhan bets, terutama pada bets