FH-Freiburg: a novel missense mutation (C317Y) in growth factor
repeat A of the low density lipoprotein receptor gene in a German
patient with homozygous familial hypercholesterolemia
Markus S. Nauck
a,*, Hubert Scharnagl
a, Henrik Nissen
b, Christine Schu¨rmann
a,
Dirk Mattern
c, Matthias A. Nauck
a, Heinrich Wieland
a, Winfried Ma¨rz
aaDepartment of Clinical Chemistry,Uni6ersity Hospital,Hugstetter Strasse 55,79106 Freiburg, Germany bDepartment of Clinical Chemistry,Uni6ersity Hospital,Odense, Denmark
cDepartment of Pediatrics,Uni6ersity Hospital,Freiburg, Germany
Received 3 June 1999; received in revised form 13 September 1999; accepted 29 September 1999
Abstract
We describe the characterization of a novel mutation in the low density lipoprotein receptor (LDL-R) gene in a patient with true homozygous familial hypercholesterolemia (FH). The combined use of denaturing gradient gel electrophoresis (DGGE) and sequencing of genomic DNA revealed a guanine to adenine base substitution at nucleotide position 1013 of the LDL-R cDNA. This point mutation results in a change from cysteine to tyrosine at amino acid residue 317 of repeat A of the epidermal growth factor (EGF) precursor homology domain. Binding, uptake and degradation of iodinated LDL in skin fibroblasts from the homozygous patient were less than 10% of normal. In contrast, binding, uptake and degradation of iodinated VLDL was reduced by only 60, 30, and 38%, respectively. Incubation of the patient’s fibroblasts in the presence of cholesterol diminished the residual binding of VLDL by 50%, suggesting that the loss of the highly conserved cysteine at position 317 results in a LDL-R that fails to bind LDL, but retains some ability to bind VLDL by interacting with the apolipoprotein E. Both parents were heterozygous for the C317Y mutation. Interestingly, however, the father presented with markedly elevated levels of triglycerides and VLDL cholesterol, whereas his LDL cholesterol was unexpectedly low. The mother of the index patient had only slightly elevated LDL cholesterol. These observations testify to the biological complexity of genotype-environment interactions in individuals carrying mutations at the LDL-R locus and indicate that genetic analysis importantly complements the clinical and biochemical diagnosis of patients with hyperlipidemia. © 2000 Elsevier Science Ireland Ltd. All rights reserved.
Keywords:Familial hypercholesterolemia; Low density lipoprotein receptor; Genetic disease; Polymerase chain reaction; Denaturing gradient gel electrophoresis; Low density lipoprotein binding
www.elsevier.com/locate/atherosclerosis
1. Introduction
Familial hypercholesterolemia (FH) is an autosomal dominant inherited disease caused by mutations in the gene coding for the low density lipoprotein receptor
(LDL-R). The LDL-R mediates the specific uptake and catabolism of LDL into the liver and many other tissues of the body [1]. Heterozygous carriers of one defective LDL-R gene express only half the number of functional LDL receptors and have a markedly raised plasma cholesterol that is frequently associated with the
occurrence of tendon xanthomata, accelerated
atherosclerosis, and premature coronary artery disease. Homozygous individuals are more severely affected and may die from CHD before reaching the age of maturity.
The prevalence of heterozygous FH appears to be approximately 0.2% in European and North American populations [2]. In some culturally and geographically
Abbre6iations: Apo, apolipoprotein; DGGE, denaturing gradient
gel electrophoresis; EDTA, ethylenediaminetetraacetate; PCR, poly-merase chain reaction; PID, pedigree identification number; RFLP, restriction fragment length polymorphism; VLDL, LDL, HDL, very low, low and high density lipoproteins; VLDL-chol, LDL-chol, HDL-chol, cholesterol of VLDL, LDL and HDL.
* Corresponding author. Tel.: +49-761-270-3779; fax: + 49-761-270-3444.
E-mail address:[email protected] (M.S. Nauck)
M.S.Nauck et al./Atherosclerosis151 (2000) 525 – 534 526
isolated population groups, however, the frequency of the disease and of specific mutations is much higher, presumably as a result of a founder effect; examples include the French Canadians [3], Sephardic Jews [4], Lebanese Christian Arabs [5], South African Afrikaners [6] and the Finns [7].
So far, more than 500 different mutant alleles of the LDL-R gene have been reported [2,8 – 10] (website: www.ucl.ac.uk/fh), and except for the few populations dominated by founder mutations, each family is, a priori, expected to have a unique LDL-R mutation. For a number of mutations identified so far, the mutant LDL-R protein has been characterized, either in cul-tured cells derived from the patient or by mutagenesis and expression of the mutant allele in vitro.
Because the LDL-R consists of distinct structural domains, different mutations result in mutant proteins whose structure and function are impaired in different ways and to a different extent. Defining mutations at the protein level allows the distinction of five classes of functional defects. These classes include defects in syn-thesis, intracellular transport, ligand binding, internal-ization, and recycling of the receptor [2,8]. Co-nsequently, the characterization of the specific mutation in the LDL-R gene of an FH patient not only provides further insights in the way the LDL-R functions in vivo, but also allows an accurate diagnosis to be made on which treatment and counselling can be based. It has long been recognized that there is considerable variation in the severity of the disease in FH patients,
in both the degree of their hypercholesterolemia and the age of onset of clinical symptoms of coronary heart disease. Comparing groups of patients with either the same or different mutations in the LDL-R gene may allow a better assessment of the underlying genetic or environmental causes of this variation. In addition, there is an ongoing debate whether or not the knowl-edge of the molecular defect may allow to better predict the response to treatment [11 – 14].
In this report we describe the identification of a novel mutation in the LDL-R gene in a FH family of German origin. The molecular defect consists of a G to A transition at nucleotide position 1013 in exon 7, which results in an amino acid substitution of cysteine by tyrosine at position 317. The index patient is a true homozygote for this mutation, thus affording the rare opportunity to unequivocally study the phenotypic ef-fects of that particular mutation. The consanguineous parents as well as two siblings are heterozygous carriers of the mutant allele and show considerable variations in the biochemical expression of the genetic defect.
2. Methods
2.1. Subjects
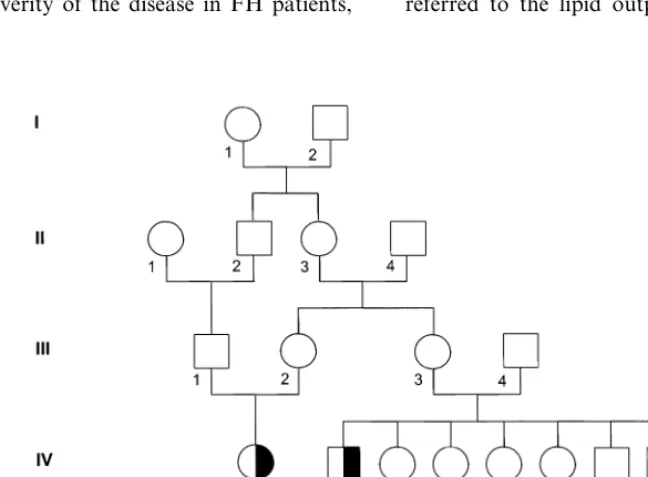
The index patient (Fig. 1, PID V-2) described in this study is a now 11-year-old German boy who was referred to the lipid outpatient clinic because of
thomas on the proximal interphalangeal joints of the fingers and bilateral lipoid arcs. No tendon xanthomas were observed. The boy had no angina, but the elec-trocardiogram showed discrete repolarisation abnor-malities in the anterior leads. Initial lipoprotein analysis showed total cholesterol of 27.5, HDL-chol of 1.0, LDL-chol of 25.9, VLDL-chol of 0.6 and triglyce-rides of 1.9 mmol/l, respectively. The index patient has one sister and one brother, born in 1985 and 1982, respectively, who are both in good health. The parents of the index patient (Fig. 1, PID IV-1 and -2) are first degree cousins, as their mothers were sisters. In addi-tion, as the parents of the index patient’s mother were first cousins as well, the extent of consanguinity in this family is extremely high. Unfortunately, only the par-ents and siblings of the index patient were available for DNA studies and analysis of serum lipid levels.
2.2. Biochemical analysis
Plasma lipoproteins were separated by preparative ultracentrifugation and precipitation and cholesterol and triglycerides were assayed by enzymatic methods
(Roche Diagnostics, Mannheim, Germany) [15].
Plasma A-I and B apolipoproteins were measured by immunonephelometry (Behringwerke AG, Marburg, Germany). Lp (a) was determined with polyclonal an-tibodies (Inkstar, Stillwater, MN) on a Behring neph-elometer as described in detail by Nauck et al. [16]. Apo E phenotyping was performed by agarose gel by isoelectric focusing and immunofixation as described previously [17].
2.3. DNA isolation and Southern blot analysis
High molecular weight genomic DNA was extracted from peripheral leukocytes according to standard pro-cedures [18]. Southern blot analysis of the LDL-R gene was performed essentially as described by
Ru¨di-ger et al. [19]. DNA (10 mg) was digested using 10
U/mg of BglII or BamHI under conditions
recom-mended by the manufacturer (New England Biolabs, Beverly, MA), separated by agarose gel electrophoresis and transferred to nylon membranes (Hybond-N, Amersham International, Buckinghamshire, UK). Two cDNAs were used for hybridization: a 1.7 kb HindIII/
BglII fragment containing sequences from exons 1
through 11 and a 1.9 kbBamHI fragment
encompass-ing sequences from exons 11 through 18 of the cDNA clone pLDLR3 (American Type Culture Collection, Manassas, VA).
2.4. Haplotype analysis of the LDL receptor locus
The haplotypes of the FH-family members were de-termined by seven known polymorphisms in the
LDL-R gene. The variable restriction sites, SfaNI in exon 2 [20],StuI in exon 8 [21],AciI in exon 11 [22], HincII in exon 12 [23], A6aII in exon 13 [24], MspI in exon
15 [25] and NcoI in exon 18 [26] were detected
by PCR amplification and restriction enzyme diges-tion. Alleles were designated as ‘+’ or ‘−’ indicating the presence or absence, respectively, of the cutting sites.
2.5. Polymerase chain reaction and DGGE
To screen for small deletions, insertions or point mutations in the LDL-R gene and in the sequence of the apolipoprotein B-100 gene encompassing the do-main relevant for receptor binding, denaturing gradi-ent gel electrophoresis (DGGE) was applied according to Nissen et al. [27]. Briefly, genomic DNA encoding the LDL-R was amplified using intron-specific primers for each of the 18 exons encompassing the coding and the splice site consensus sequences at the intron/exon junctions. The gene regions screened by the exon 1 and LDL-R promoter primers are overlapping and in combination screen all of exon 1 and the first 233 bp of the LDL-R promoter sequences, thus including all the known regulatory elements of the LDL-R gene [28]. With the primers specific for the apo B gene only codons 3456 – 3553 are screened, but these include all known mutations resulting in defective binding of apo B to the LDL-R [29].
2.6. Sequencing of exon 7 of the LDL receptor gene
PCR fragments of exon 7 that displayed an abnor-mal pattern in DGGE were purified by glass milk elution of the appropriate band from 2% agarose (Jet Quick, Genomed GmbH, Bad Oeynhausen, Germany) and ethanol precipitation. The purified fragments were sequenced directly in both directions using the fluores-cence-labeled primers SP 66 and 67 [8] and the thermo sequenase cycle sequencing kit (US Biochemical, Cleveland, OH) according to the manufacturer’s in-structions. Aliquots of the sequencing reactions were then analyzed using an automated sequencer (ALF, Pharmacia, Uppsala, Sweden).
2.7. Restriction analysis of the C317Y mutation
Because the GA transition at nucleotide position
M.S.Nauck et al./Atherosclerosis151 (2000) 525 – 534 528
2.8. Binding, uptake and degradation of125I-labeled
lipoproteins
VLDL (dB1.0063 kg/l) and LDL (1.019BdB1.063
kg/l) were isolated by preparative ultracentrifugation from a pool of normolipemic donors and iodinated using the iodine monochloride method [30]. Human skin fibroblasts were obtained from skin biopsies of the patient and a normolipemic individual, respectively. Cells were grown in 24-well polystyrene plates. Prior to the experiments, cells were pre-incubated for 40 h in
medium containing 10% (v/v) human
lipoprotein-defi-cient serum in order to up-regulate LDL receptors.
Binding, uptake and degradation of 125
I-labeled lipo-proteins were measured according to the procedure described by Goldstein et al. [31] with slight modifica-tions [32]. To measure cell surface binding, cells were incubated for 1 h at 4°C with 125I-labeled lipoproteins
in DMEM medium containing 10 mM HEPES. To determine uptake (surface binding plus internalization) and degradation, cells were incubated for 4 h at 37°C
with 125I-labeled lipoproteins in DMEM medium
con-taining 24 mM bicarbonate (pH 7.4). The amount of
125I-labeled material associated with the cells (binding
and internalization) was determined as 125
I-labeled trichloracetic acid-soluble (non-iodine) material in the conditioned medium. Values were corrected for protein concentrations using the Lowry assay and BSA as standards (Bio-Rad, Hercules, CA).
3. Results
3.1. Family analysis
The pedigree of the G. family is shown in Fig. 1 and the plasma lipid values are given in Table 1.
The parents of the patient were first degree cousins, as the parents’ mothers were sisters (PID III-2 and -3). Further, the maternal grandparents of the patient were also first degree cousins (PID III-1 and -2), so that an extraordinary extent of consanguinity is evident in this family. The patient showed lipid values that definitively met the biochemical criteria for homozygous FH. The patient presented with xanthomas on the proximal in-terphalangeal joints of the fingers and bilateral lipoid arcs characteristic of FH. On ECG, discrete repolarisa-tion abnormalities were detected, which we interpret as an early sign of ischemic cardiomyopathy. The patient
is now treated with colestyramine (16 g/d) and
LDL-apheresis which is performed every 2 weeks.
Both siblings of the patient also showed concentra-tions of LDL-chol that were above the 95th percentile for age and gender in the German population. Both siblings were doing well and had no clinical signs of
FH. The mother showed moderate
hypercholes-terolemia only with LDL-chol and HDL-chol concen-trations that were both above the 75th percentile for age and sex. Fasting triglycerides, however, were signifi-cantly elevated.
Of special interest was the biochemical analysis of the patient’s father, who showed an extreme fasting hyper-trigyceridemia and an unusually high VLDL-chol con-centration. The concentration of LDL-chol, however, was 70 mg/dl, thus being below the 5th percentile of age — and sex adjusted reference values. Consequently, at a glance, the presence of a mutated LDL-R allele appeared to be rather unlikely in this individual. Both parents showed no clinical signs of FH. The self-re-ported alcohol consumption of the parents was 150 and 600 g ethanol per week for mother and father, respec-tively. The g-glutamyl-transferase activity of the father was elevated at 87 U/l (local reference range: 0 – 24 U/l). Unfortunately, because the patient’s parents have completely lost contact with their relatives, further members of the G. family were not available for clinical examination, DNA testing, or biochemical analyses. Information on the causes of death in previous genera-tions was also not obtainable.
3.2. Identification of a sequence alteration by DGGE
Southern blot analysis after BglII or XbaI digestion of genomic DNA isolated from the patient, his parents and his siblings did not reveal any major abnormality in the LDL-R gene (not shown), suggesting the pres-ence of a point mutation or minor rearrangement. To determine the exact location of the mutation, the am-plified products of all 18 exons, including the splice site sequences, and of the promoter of the LDL-R of the patient, his parents and his siblings were subjected to DGGE analysis.
As shown in Fig. 2, evidence for a single mutation was found in exon 7 or in its splice junctions.
The DGGE analysis for the patient revealed a single homoduplex band as it is seen in individuals with two identical alleles. However, the electrophoretic mobility of this homoduplex band was clearly distinct from that of two normolipemic controls. The mutant homoduplex band migrated a shorter distance in the denaturing gradient gel, indicating a lower melting temperature of the altered sequence. The appearance of this single homoduplex band with an aberrant melting profile clearly indicated a true homozygous state with the presence of an identical mutation on both alleles.
.
S
.
Nauck
et
al
.
/
Atherosclerosis
151
(2000)
525
–
534
529
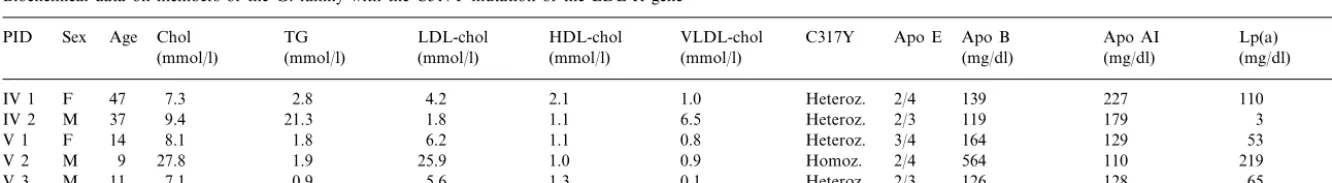
Table 1
Biochemical data on members of the G. family with the C317Y mutation of the LDL-R genea
TG LDL-chol
Chol HDL-chol
Age VLDL-chol
Sex C317Y Apo E
PID Apo B Apo AI Lp(a)
(mg/dl) (mg/dl)
(mmol/l) (mmol/l) (mmol/l) (mg/dl)
(mmol/l) (mmol/l)
2.1
IV 1 F 47 7.3 2.8 4.2 1.0 Heteroz. 2/4 139 227 110
1.1 6.5
IV 2 M 37 9.4 21.3 1.8 Heteroz. 2/3 119 179 3
1.1 0.8 Heteroz. 3/4 164 129
6.2 53
8.1 1.8
V 1 F 14
25.9
M 9 27.8 1.9 1.0 0.9 Homoz. 2/4 564 110 219
V 2
0.9 5.6 1.3 0.1 Heteroz. 2/3 126 128 65
V 3 M 11 7.1
M.S.Nauck et al./Atherosclerosis151 (2000) 525 – 534 530
Fig. 2. DGGE analysis of the patient and his family. Exon 7 was amplified from genomic DNA as described in Section 2. The PCR product, containing a 40-bp GC clamp was loaded on a 6% poly-acryl-amide gel containing a denaturing gradient of 40 – 80%. Pedigree identification number is with reference to Table 1. A single homodu-plex band with an abnormal electrophoretic mobility, indicating the presence of an identical mutation on both alleles is seen in subject V2. All other family members examined display an identical four band pattern with a fast-migrating homoduplex band at the position of the normolipemic controls (lane 6 and 7) and a slow-migrating homodu-plex band at the position corresponding to the mutant DNA.
bonds that are located in repeat A of the EGF precur-sor homology domain of the LDL-R protein [2].
As the base substitution at position 1013 creates a
recognition sequence of RsaI (GTAC), the presence of
the mutation was confirmed by RsaI digestion of the
PCR product of exon 7. As expected, the 253 bp PCR product originating from the patient was completely digested, generating two fragments with 146 and 107 bp, respectively. The PCR products of the controls remained completely uncut, whereas the other family members displayed restriction fragments consistent with a heterozygous state for this mutation (not shown).
3.4. Haplotype analysis
Genotyping at seven polymorphic sites showed that the index patient was homozygous for the SfaNI (+),
StuI (+), AciI (+), HincII (−), A6aII (+), MspI
(+) and the NcoI (−) RFLPs. This genotype was
identical for all family members except for the NcoI
polymorphic site, at which the mother and the sister of the patient were heterozygous.
3.5. Interaction of lipoproteins with cultured cells
To examine the functional properties of the mutant LDL-R, skin biopsies of the patient and a nor-molipemic individual were obtained and primary fibroblast cell cultures were established. We studied binding (at 4°C), uptake and degradation (both at 37°C) of125I-LDL and125I-VLDL at protein
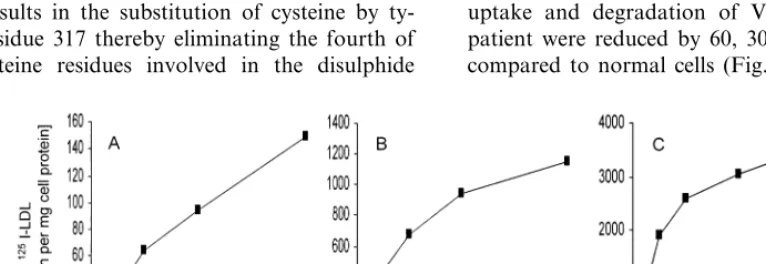
concentra-tions of 5 through 40 mg/l. Binding, uptake and degra-dation of125I-LDL in fibroblasts from the patient were
less than 10% of those in normal cells (Fig. 3). The interaction of 125I-VLDL with cultured cells was also
decreased, but to a lesser extent. On average, binding, uptake and degradation of VLDL in cells from the patient were reduced by 60, 30, and 38%, respectively, compared to normal cells (Fig. 4).
the same position as the homoduplex band of the controls, representing the normal allele whereas the second lowest band displayed the same elctrophoretic mobility as the homoduplex band seen in the patient’s sample.
3.3. Nucleotide sequence of the mutant allele and
restriction genotyping of the C317Y mutation
To precisely identify the mutation in exon 7, the PCR product originating from the mutant allele of the pa-tient was sequenced. Direct bidirectional sequencing
revealed a single base substitution (GA) at
nucle-otide 1013 of the LDL-R cDNA (not shown). This mutation results in the substitution of cysteine by ty-rosine at residue 317 thereby eliminating the fourth of the six cysteine residues involved in the disulphide
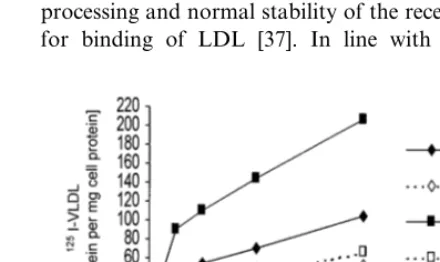
Fig. 4. Binding, uptake and degradation of125I-VLDL. VLDL were prepared by ultracentrifugation and labelled with125I as described in Section 2. Skin fibroblasts were grown in DMEM with 10% (v/v) FCS. Prior to the experiment (40 h), they were switched to medium containing 10% (v/v) human lipoprotein-deficient serum. Cells from the patient (rhombs) and from a normolipemic donor (squares) then received125I-labeled VLDL. Binding (panel A), uptake (panel B), and degradation (panel C) were determined as described Section 2. Each data point represents the average of two experiments, each performed in triplicate.
Binding of VLDL was also determined after down-regulating the expression of the LDL-R gene by incu-bating the cells for 24 h in the presence of sterols [33]. In the cells of the patient, sterol-dependent repression of the LDL-R gene diminished the residual binding activity for VLDL by 50% (Fig. 5).
4. Discussion
A missense mutation in the LDL-R gene has been detected in a homozygous FH patient with the com-bined use of DGGE and sequence analysis. The mutant alleles inherited by the patient from both parents change the codon 317 from TGC to TAC. This transi-tion results in a replacement of cysteine by tyrosine at position 317 of the LDL-R protein, thereby eliminating the fourth of six cysteine residues involved in the for-mation of disulphide bonds located in repeat A of the EGF precursor homology domain of the LDL-R [28]. This domain is the most highly conserved in evolution, and the cysteine residues in repeat A are absolutely conserved [34]. The mutation reported here is the sec-ond mutation affecting codon 317. A previous report described a homozygous FH patient from Japan with a transition from TGC to TAC [35]. That mutation, named FH-Wakayama, causes an amino acid change from cysteine to serine. In pulse chase experiments performed with fibroblasts from the Japanese ho-mozygous patient, Funahashi et al. found that the
posttranslational processing of the receptor was
markedly impaired, and the majority of the receptors remained in the precursor form even after the chase period [36]. Accordingly, the allele FH-Wakayama en-coded a protein that is delayed in transport between the endoplasmic reticulum and the Golgi apparatus. Al-though the mutation described in this paper is not a serine for cysteine317, but a tyrosine for cysteine317
substitution, it is reasonable to assume that the func-tional consequences are similar, because a disulphide bond is lost in both cases. Thus, also in view of the similarity between serine and tyrosine, it is likely that a defect in the intracellular transport of the receptor is present in our patient as well, so that we propose to classify the newly discovered allele C317Y as transport defective, the most common type of mutation at the LDL-R locus [8].
In a previous study using site-directed mutagenesis, Esser et al. showed that repeat A of the EGF precursor homology region is required not only for the regular processing and normal stability of the receptor, but also for binding of LDL [37]. In line with this previous
M.S.Nauck et al./Atherosclerosis151 (2000) 525 – 534 532
observation, the LDL-R assay of our patient’s fibrob-lasts revealed that 125I-LDL binding, internalization
and degradation were all markedly reduced to values that are compatible with receptor-negative homozygous FH [1]. Similar values have been obtained with cells from the homozygous patient with the FH-Wakayama mutation [36]. Extending the functional characteriza-tion of Funahashi et al. we also performed binding studies using apo E containing125I-VLDL as ligand, the
binding of which to the LDL-R has less stringent structural requirements than the binding of LDL [38]. We found that binding of VLDL was reduced to only about half the normal value. In addition, when the expression of the LDL-R gene was repressed by incu-bating the cells in the presence of sterols, the mutant cells showed a residual binding activity for VLDL that was further diminished by 50%, compared to cells cultured under sterol-depleted conditions. These results suggest that half of the residual binding of VLDL seen in sterol-depleted cells was as a result of the LDL-R and that the proportion of the mutant receptor that reaches the cell surface selectively looses the ability to
bind 125I-LDL, but retains an ability to bind 125
I-VLDL. This assumption is supported by experiments from Esser et al. showing that the replacement of either 1 or 2 cysteine residues in repeat A (cysteine297 and
cysteine308) selectively decreases binding of LDL, but
not of b-VLDL [37]. Taken together, the mutation
C317Y presumably encodes a receptor protein that exhibits more than one type of functional defect. It is converted to the mature form at a very slow rate, and the reduced amounts of LDL-R reaching the cell sur-face specifically fails to bind LDL, but not VLDL. Accordingly, the type of mutation encoded by this allele has to be classified as class 2b mutation [8].
Further evidence that the mutation C317Y is causing FH comes from the family analysis. The homozygous patient is more severely affected than his two older siblings, who are heterozygous for the C317Y allele, so that a gene-dosage effect typical of autosomal-codomi-nant disorders can be established. While both siblings had an intermediate level of LDL-chol consistent with the presence of only a single functional LDL-R gene, the family analysis turned more complicated when tak-ing the parents into account. The patient’s mother showed concentrations of total cholesterol and triglyce-rides above the 95th percentile for age and gender, while LDL-chol was only moderately elevated being above the 75th percentile. HDL-chol was elevated (2.1 mmol/l) above the 75th percentile as well. The father showed an extremely elevated triglyceride level (20.7 mmol/l), while his LDL-chol concentration was strik-ingly low.
It is known that many genetic and environmental factors influence the LDL-chol so that a reasonable number of individuals is misclassified as having or not
having FH when traditional clinical criteria are used to make the diagnosis. According to the probability as-sessments of Williams et al. for diagnosing FH in members of FH families, the mother and the father of the patient had probabilities of 11.2% and below 1%, respectively, for having FH if LDL-chol levels were used and it is assumed that a first degree relative has definite FH [39]. Although Williams et al. suggested using lower diagnostic lipid values in FH families than in the general population, based on the higher a priori possibility of having FH when belonging to an FH family than to the general population, these criteria would fail to diagnose the correct FH status of the parents in this case. In case of the father one would even tend to exclude the presence of a defective LDL-R allele because of the extremely low LDL-chol level. In addition, clinical symptoms of FH, such as tendon xanthomas or evidence for the presence of CAD were absent in both parents. These low probabilities on the basis of biochemical and clinical criteria, despite the presence of a disease-causing LDL-R allele, document the improved diagnostic precision obtained by intro-ducing genetic diagnosis in FH families [40 – 42].
What are the factors that mitigate the hypercholes-terolemic phenotype of the parents despite the presence of a disease-causing allele that clearly results in a characteristic FH-phenotype in the affected children? In our opinion, the uncharacteristic phenotype of the par-ents is most likely attributable to their nutritional be-haviour, which is characterized by both strikingly hypercaloric diet and high alcohol consumption, both of which being present to a greater extent in the father.
Ethanol ingestion is known to affect the lipid
metabolism by stimulating the synthesis of VLDL triglycerides in the liver. In addition it is discussed that alcohol delays the metabolism of VLDL resulting in a decreased production rate of LDL [43,44]. Moreover, extreme alcohol consumption may also contribute to a reduction in LDL-chol, because of the low content of unsaturated fatty acids typically present in the diet of these individuals [45]. Here, it seems conceivable that the decreased metabolism of LDL as a result of the presence of a functionally defective LDL-R is compen-sated by an impaired lipolysis of VLDL resulting in a reduced production rate of LDL. As a consequence, the LDL steady state concentrations in plasma are not profoundly elevated, as found in the mother, or even unexpectedly low, as seen in the father.
In line with the well established positive correlation between alcohol consumption and HDL, the mother showed a notably elevated concentration of HDL-chol [44,46].
at the University Hospital Freiburg, we did not detect any further carrier of the mutation C317Y. In view of the patient’s family exhibiting first degree consanguinity in two generations, one can speculate that both alleles affected by the same mutation present in the ho-mozygous patient are derived from the same ancestor. This view is supported by the haplotype analysis show-ing homozygosity for all polymorphic sites tested. The family of the patient is of South-West German origin, and we propose that this mutation should be named FH-Freiburg according to the nomenclature of Hobbs et al. [2].
In conclusion, the mutant allele identified in the homozygous patient encodes a LDL-R that is defective for both transport and LDL binding. The allele is associated with a severe clinical phenotype in the ho-mozygous patient and his heterozygous siblings. The presentation of the heterozygous parents, however, un-derlines the profound impact of life-style habits, which might blur the diagnosis of FH based on clinical crite-ria. These uncertainties seen in the parents call for simple and efficient methods for genetic diagnosis in order to improve diagnostic precision [40,47].
Acknowledgements
The excellent technical assistance of Ulrike Stein, Sabine von Karger, Sybille Rall and Brigitte Kreisel is gratefully acknowledged.
References
[1] Goldstein JL, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Diseases, 7th ed. New York: Mc-Graw-Hill, 1995:1981.
[2] Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational anal-ysis of a membrane protein. Annu Rev Genet 1990;24:133. [3] Leitersdorf E, Tobin EJ, Davignon J, Hobbs HH. Common
low-density lipoprotein receptor mutations in the French Cana-dian population. J Clin Invest 1990;85:1014.
[4] Leitersdorf E, Reshef A, Meiner V, Dann EJ, Beigel Y, van Roggen FG, van der Westhuyzen DR, Coetzee GA. A missense mutation in the low density lipoprotein receptor gene causes familial hypercholesterolemia in Sephardic Jews. Hum Genet 1993;91:141.
[5] Lehrman MA, Schneider WJ, Brown MS, Davis CG, Elhammer A, Russell DW, Goldstein JL. The Lebanese allele at the low density lipoprotein receptor locus. Nonsense mutation produces truncated receptor that is retained in endoplasmic reticulum. J Biol Chem 1987;262:401.
[6] Leitersdorf E, van der Westhuyzen DR, Coetzee GA, Hobbs HH. Two common low density lipoprotein receptor gene muta-tions cause familial hypercholesterolemia in Afrikaners. J Clin Invest 1989;84:954.
[7] Koivisto UM, Turtola H, Aalto-Setala K, Top B, Frants RR, Kovanen PT, Syvanen AC, Kontula K. The familial
hyperc-holesterolemia (FH)-North Karelia mutation of the low density lipoprotein receptor gene deletes seven nucleotides of exon 6 and is a common cause of FH in Finland. J Clin Invest 1992;90:219. [8] Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1992;1:445.
[9] Varret M, Rabes JP, Thiart R, Kotze MJ, Baron H, Cenarro A, Descamps O, Ebhardt M, Hondelijn JC, Kostner GM, Miyake Y, Pocovi M, Schmidt H, Schuster H, Stuhrmann M, Yama-mura T, Junien C, Beroud C, Boileau C. Ldlr Database (2nd ed.) — new additions to the database and the software, and results of the first molecular analysis. Nucleic Acids Res 1998;26:248. [10] Wilson DJ, Gahan M, Haddad L, Heath K, Whittal RA,
Williams RR, Humphries SE, Day INM. A world wide web site for low-density lipoprotein receptor gene mutations in familial hypercholesterolemia — sequence-based, tabular, and direct submission data handling. Am J Cardiol 1998;81:1509. [11] Jeenah M, September W, Graadt van Roggen F, de Villiers W,
Seftel H, Marais D. Influence of specific mutations at the LDL-receptor gene locus on the response to simvastatin therapy in Afrikaner patients with heterozygous familial hypercholestero-laemia. Atherosclerosis 1993;98:51.
[12] Leitersdorf E, Eisenberg S, Eliav O, Friedlander Y, Berkman N, Dann EJ, Landsberger D, Sehayek E, Meiner V, Wurm M. Genetic determinants of responsiveness to the HMG-CoA reduc-tase inhibitor fluvastatin in patients with molecularly defined heterozygous familial hypercholesterolemia. Circulation 1993;87:III35.
[13] Sun XM, Patel DD, Knight BL, Soutar AK. Influence of genotype at the low density lipoprotein (Ldl) receptor gene locus on the clinical phenotype and response to lipid-lowering drug therapy in heterozygous familial hypercholesterolaemia. Atherosclerosis 1998;136:175.
[14] Sijbrands EJ, Lombardi MP, Westendorp RG, Leuven JA, Meinders AE, Vanderlaarse A, Frants RR, Havekes LM, Smelt AH. Similar response to simvastatin in patients heterozygous for familial hypercholesterolemia with mRNA negative and mRNA positive mutations. Atherosclerosis 1998;136:247.
[15] Program LRC. Lipid and Lipoprotein Analysis, 2nd ed. Bethesda: NIH, 1982.
[16] Nauck M, Winkler K, Wittmann C, Mayer H, Luley C, Ma¨rz W, Wieland H. Direct determination of lipoprotein(a) choles-terol by ultracentrifugation and agarose gel electrophoresis with enzymatic staining for cholesterol. Clin Chem 1995;41:731. [17] Luley C, Baumstark MW, Wieland H. Rapid apolipoprotein E
phenotyping by immunofixation in agarose. J Lipid Res 1991;32:880.
[18] Miller SA, Dykes DD, Polesky HF. A simple salting out proce-dure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215.
[19] Ru¨diger NS, Heinsvig EM, Hansen FA, Faergeman O, Bolund L, Gregersen N. DNA deletions in the low density lipoprotein (LDL) receptor gene in Danish families with familial hyperc-holesterolemia. Clin Genet 1991;39:451.
[20] Soutar AK. A polymorphism in exon 2 of the human LDL-re-ceptor gene (LDLR). Nucleic Acids Res 1991;19:4314. [21] Kotze MJ, Langenhoven E, Warnich L, Marx MP, Retief AE.
Molecular characterisation of a low-frequency mutation in exon 8 of the human low-density lipoprotein receptor gene. S Afr Med J 1989;76:402.
M.S.Nauck et al./Atherosclerosis151 (2000) 525 – 534 534
[24] Hobbs HH, Esser V, Russell DW.A6aII polymorphism in the
human LDL receptor gene. Nucleic Acids Res 1987;15:379. [25] Geisel J, Weisshaar B, Oette K, Mechtel M, Doerfler W. Double
MspI RFLP in the human LDL receptor gene. Nucleic Acids Res 1987;15:3943.
[26] Kotze MJ, Langenhoven E, Dietzsch E, Retief AE. A RFLP associated with the low-density lipoprotein receptor gene (LDLR). Nucleic Acids Res 1987;15:376.
[27] Nissen H, Guldberg P, Hansen AB, Petersen NE, Horder M. Clinically applicable mutation screening in familial hypercholes-terolemia. Hum Mutat 1996;8:168.
[28] Sudhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science 1985;228:815.
[29] Nissen H, Hansen PS, Faergeman O, Horder M. Mutation screening of the codon 3500 region of the apolipoprotein B gene by denaturing gradient-gel electrophoresis. Clin Chem 1995;41:419.
[30] Goldstein JL, Brown MS. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with ho-mozygous familial hypercholesterolemia. J Biol Chem 1974;249:5153.
[31] Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocy-tosis of low-density lipoprotein in cultured cells. Methods Enzy-mol 1983;98:241.
[32] Ma¨rz W, Baumstark MW, Scharnagl H, Ruzicka V, Buxbaum S, Herwig J, Pohl T, Russ A, Schaaf L, Berg A, Gross W. Accumu-lation of ‘small dense’ low density lipoproteins (LDL) in a homozygous patients with familial defective apolipoprotein B-100 results from heterogenous interaction of LDL subfractions with the LDL receptor. J Clin Invest 1993;92:2922.
[33] Goldstein JL, Brown MS. Regulation of the mevalonate path-way. Nature 1990;343:425.
[34] Mehta KD, Chen WJ, Goldstein JL, Brown MS. The low density lipoprotein receptor inXenopus lae6is. I. Five domains
that resemble the human receptor. J Biol Chem 1991;266:10406. [35] Funahashi T, Yamashita S, Maruyama T, Ueyama Y, Menju M, Nagai Y, Takemura K, Miyake Y, Tajima S, Matsuzawa Y. A compound heterozygote for familial hypercholesterolaemia with a homozygous mother. J Intern Med 1996;239:187.
[36] Funahashi T, Miyake Y, Yamamoto A, Matsuzawa Y, Kishino B. Mutations of the low density lipoprotein receptor in Japanese
kindreds with familial hypercholesterolemia. Hum Genet 1988;79:103.
[37] Esser V, Limbird LE, Brown MS, Goldstein JL, Russell DW. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J Biol Chem 1988;263:13282. [38] Russell DW, Brown MS, Goldstein JL. Different combinations
of cysteine-rich repeats mediate binding of low density lipo-protein receptor to two different lipo-proteins. J Biol Chem 1989;264:21682.
[39] Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, Hopkins PN. Diagnosing heterozygous famil-ial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol 1993;72:171.
[40] Nissen H, Hansen AB, Guldberg P, Petersen NE, Larsen ML, Haghfelt T, Kristiansen K, Horder M. Genetic diagnosis with the denaturing gradient gel electrophoresis technique improves diagnostic precision in familial hypercholesterolemia. Circulation 1995;91:1641.
[41] Koivisto UM, Hamalainen L, Taskinen MR, Kettunen K, Kon-tula K. Prevalence of familial hypercholesterolemia among young north Karelian patients with coronary heart disease: a study based on diagnosis by polymerase chain reaction. J Lipid Res 1993;34:269.
[42] Ward AJ, O’Kane M, Nicholls DP, Young IS, Nevin NC, Graham CA. A novel single base deletion in the LDLR gene (211delG): effect on serum lipid profiles and the influence of other genetic polymorphisms in the ACE, APOE and APOB genes. Atherosclerosis 1996;120:83.
[43] Chait A, Mancini M, February AW, Lewis B. Clinical and metabolic study of alcoholic hyperlipidaemia. Lancet 1972;2:62. [44] Taskinen MR, Nikkila EA, Valimaki M, Sane T, Kuusi T, Kesaniemi A, Ylikahri T. Alcohol-induced changes in serum lipoproteins and in their metabolism. Am Heart J 1987;113:458. [45] Hulley SB, Gordon S. Alcohol and high-density lipoprotein cholesterol: causal inference from diverse study designs. Circula-tion 1981;64:III57.
[46] Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, Kagan A, Zukel WJ. Alcohol and blood lipids. The cooperative lipoprotein phenotyping study. Lancet 1977;2:153. [47] Schuster H, Luft FC. Clinical criteria versus DNA diagnosis in
heterozygous familial hypercholesterolemia — is molecular diag-nosis superior to clinical diagdiag-nosis? Arterioscler Thromb Vasc Biol 1998;18:331.