U N I VERTAS
UDAYANA,iRUMAH
SAICT
TJMIJMPUSAT
SANGLAH
DENPASARJlrr P. Serangan Dps. Bali(80114) Email. [email protected] Telp. (0361) 221453 4, (0361)227911-15(p.227)
Nomor
:((26
nsr't.t+.2Litban{2015Lampiran
:
1 lembarPerihal
:
Penyerahan Elftical ClemcmceKepada
Yth.
\
Dr.dr. Made Ratna Saraswati. SpPD-KEMD di-Tempat
Dengan hormat,
Bersama ini kami menyerahkan Erft ical Clearance lKeterangan Kelaikan Etik Nomor : 1800ruN. 14.2l1,itban!2015, tsrtanggal, 26 Nopember 2015
Hal-hal yang perlu diperhatikan :
1.
Setelah selesai penelitian wajib menyeralkan 1 (satu) copy hasil penelitiannya.2.
Jika ada perubahan yang menyangkut dengan hal penelitian tersebut mohon melaporkan ke Komisi Etik Litbang FK UNUDIRSUP Sanglah Denpasar.Demikian kami sampaikan, atas perhatian dan keriasamanya kami ucapkan terimakasih. Denpasar,
3 ^
/?
^ 2Ptf
19530131't980031 Tembusan:
Arsip.-UNTVERTAS
UDAYANA/RUMAH SAKIT UMUM
PUSAT
SANGLAH
DENPASAR.lalan P. Serangan Denpasar Bali (80114) Email : l.itbang.rmud.rsup(@gmail-com Telp. (0361) 24453 4, (0361)227911-15(p.227)
ETHICAL
CLEARANCE
NO.
1 800/LrN .1 4.2/LiIbangl201 51
This is to ceftily that following study project entitled :
"DISCOWR: DISCOVERing Treatment Reality of Type 2 Diabetes in Real World Settings."
Principal Investigator : Dr.dr. Made Ratna Salaswati, SpPD-KEMD Other
Researchers
: dr. Made Pande Dwipayana, SpPD-KEMDResearch Development: RSUP Sanglah Denpasar'
Plotocol
Number
: 833.03.3-2015Has been evaluated
in accordance
with the ethical aspects in using human being as a study subject and consideled proper to be executed.1.
Progless lepoft every... month2.
Final leportNon-Interventional Study (NIS) Primary Protocol Amendment
Amendment Edition Number 1
NIS D-code DISCOVER D1690R00002
NIS Primary Protocol Dated 20 November 2014 (version 2)
Date 01 July 2015
DISCOVER: DISCOVERing Treatment Reality of Type 2 Diabetes in Real World
Settings
Sponsor:
AstraZeneca AB
151 85 Södertälje, Sweden
Financial Support By:
AstraZeneca
The following Amendment(s) have been made to this protocol since the date of preparation:
Amendment No. Date of Amendment Local Amendment No: Date of Local Amendment
Date of protocol amendment: 01 July 2015
NON-INTERVENTIONAL STUDY PRIMARY PROTOCOL SYNOPSIS
DISCOVER: DISCOVERing Treatment Reality of Type 2 Diabetes in Real
World Settings
International Co-ordinating Investigator of the Non-Interventional Study Primary
Professor Linong Ji, MD
Peking University People’s Hospital, 11 Xizhimen Nan Dajie, Xicheng District, Beijing, China
Tel. + 86 1068358517
Scientific Committee members of the Non-Interventional Study Primary
Professor Bernard Charbonnel, MD Department of Endocrinology, University of Nantes,
44000 Nantes, France Tel. + 33 (02) 40083642
Professor Marília Gomes, MD Unit of Dia betes,
Universidade Estadual do Rio de Janeiro, Avenida 28 de Setembro, 77, 3o andar, CEP 20.551-030,
Rio de Janeiro, Brazil Tel. + 55 2128688224
Professor Kamlesh Khunti, MBChB, PhD Diabetes Research Centre
University of Leicester Leicester General Hospital
Gwendolen Road, Leicester, United Kingdom LE5 4P W
Date of protocol amendment: 01 July 2015
Professor Antonio Nicolucci, MD Consorzio Mario Negri Sud,
Via Nazionale 8/a, 66030 S Maria Imbaro, Ita ly Tel.: + 39 0872570260
Professor Stuart Pocock, PhD Medical Statistics Unit,
London School of Hygiene and Tropical Medicine, Keppel Street, London, United Kingdom
WC1E 7HT
Tel. + 44 207927 2413
Professor Marina Shestakova, MD
Institute of Diabetes, Federal Scientific Centre of Endocrinology, Dmitryia Uljanova, No. 11, 117036 Moscow, Russia .
Tel. + 7 495 124 45 00
Professor Mikhail Kosiborod, MD Saint Luke's Mid America Heart Institute, University of Missouri-Kansas City, 4401 Wornall Road 600
Kansas City, MO, USATel. + 1 816 931 1883
Study Site(s), number of subjects and countries planned
Approximately 9650 subjects recruited from sites in Argentina, Australia, Austria, Brazil, Canada, China (Mainland), Colombia, Costa Rica, Czech Rep, France, Germany, India, Indonesia, Italy, Malaysia, Mexico, Netherlands, the Nordics (Denmark, Norway and Sweden) Panama, Poland, Russia, South Korea, Spain, Taiwan and the United Kingdom.
<<The list of participating countries is not yet definitive so this list is subject to change>>.
Total planned Study period
Estimated date of first subject in Q4 2014
Estimated date of last subject in Q4 2015
Estimated date of last subject last visit Q4 2018
Estimated date of data base lock Q1 2019
Medicinal Products (type, dose, mode of administration) and concomitant medication
Date of protocol amendment: 01 July 2015
Rationale for this Non-Interventional Study (NIS) Primary
In many geographical regions, data on diabetes treatments and control is scarce and where data
sources are available these do not contain enough detail to capture the entire patient journey.
Little data has been captured and published since the launch of newer classes of therapy such
as DPP-IV inhibitors, GLP-1 agonists and most recently the SGLT2 inhibitors as compared to
the well known established oral hypoglyacemic agents sulphonylureas and metformin.
In this context, this study aims to provide data on real world second and further line anti-
diabetic therapy use among type 2 diabetes patients in different geographical regions. The
association of these therapies with achieving disease control and in preventing and controlling
diabetes complications will be documented. The DISCOVER program will be the largest
global study of this kind ever performed.
Objectives of this Non-Interventional Study Primary
(a) Primary objective
The primary objective is to describe the disease management patterns and clinical
evolution over three years in type 2 diabetes mellitus patients initiating a second
line anti-diabetic treatment (add-on or switch), after a first line oral treatment with a
monotherapy, dual or triple therapy.
(b) Main secondary objective
To describe, overall and by patient characteristics as well as second line anti-
diabetic medication class, treatment response in terms of changes in HbA1c, body
weight, blood pressure and lipid profile from baseline and achievement of HbA1c
target goals.
To describe treatment changes, that is initiation of third line or above add-on anti-
diabetic medication, initiation of insulin therapy, switching of anti-diabetic
Date of protocol amendment: 01 July 2015
To describe disease progression in terms of microvascular complications, that is,
incidence of diabetic nephropathy, diabetic neuropathy, diabetic retinopathy or
diabetes-related peripheral revascularization and/or non-traumatic amputation.
To describe incidence of macrovascular complications, that is, cardiovascular death,
heart failure, myocardial infarction and stroke.
To describe hypoglycemic events and/or hospitalizations for hypoglycemia.
To describe patient reported Quality of Life.
To describe healthcare resource use.
To describe risk factors (disease, patient, treatment and setting characteristics at
baseline: e.g. age, gender, duration of diabetes, second line anti-diabetic medication
class, presence of co-morbidities, socioeconomic status, quality of care) associated
with microvascular complications, macrovascular complications, hypoglycemic
events, quality of life and healthcare resource use during follow-up.
To describe factors associated with treatment choice at baseline.
Study design
This study is a multi-country, multicenter, observational, prospective, longitudinal cohort
study. The countries patients will be recruited from are: Argentina, Australia, Austria, Brazil,
Canada, China (Mainland), Colombia, Costa Rica, Czech Rep, France, Germany, India,
Indonesia, Italy, Malaysia, Mexico, Netherlands, the Nordics (Denmark, Norway and Sweden)
Panama, Poland, Russia, South Korea, Spain, Taiwan and the United Kingdom.
<<The list of participating countries is not yet definitive so this list is subject to change>>.
It is estimated that approximately 9650 patients will be enrolled in total with each patient
followed up for 3 years.
In France, Germany, the Nordics and the United Kingdom, the observational, prospective
Date of protocol amendment: 01 July 2015
records, patient registers or disease registers. In the remaining countries, AstraZeneca and its
study partners will pursue the establishment of data networks at the sites to allow for
extraction of additional clinical information and from electronic medical records with linkage
to the NIS, in accordance with the country specific privacy laws.
Target subject population
Patients with type 2 diabetes initiating their second line anti-diabetic therapy after first line
diabetic therapy.
Study variable(s):
Primary variable
• Class of diabetic medication/s initiated as second-line anti-diabetic therapy and
associated clinical evolution (through HbA1c, body weight, blood pressure,
hypoglycemic events, micro- and macro-vascular complications).
Secondary variables
• Site characteristics.
• Physician speciality.
• Patient demographics.
• Vital signs and lab tests.
• Medical history of T2DM, including presence of risk factors.
• Co-morbidities and co-medications.
• Changes in diabetes treatments during follow-up and reasons.
• Number of major hypoglycemic events, occurrence of minor hypoglycemic events.
• Microvascular complications (nephropathy, retinopathy, neuropathy and amputation)
and macrovascular complications.
• All-cause death.
• Patient reported outcomes: Quality of Life: SF-36, HFS-II, lifestyle and healthcare
avoidance due to costs. The specific PROs to be used in each country will depend on
Date of protocol amendment: 01 July 2015
• Healthcare resource use.
Statistical methods
Statistical methods appropriate for descriptive purposes in epidemiological studies will be
used for the analysis of collected data. Interim analysis might be performed at baseline after
the last subject is recruited and then 1 and 2 years after last subject in.
It is therefore proposed to include an overall sample size of 9650 patients. This will guarantee
that, for any second line compound class given to ≥ 1.5% of patients (i.e., 201 patients),
Date: 01 July 2015
TABLE OF CONTENTS
PAGE
TITLE PAGE...1
NON-INTERVENTIONAL STUDY PRIMARY PROTOCOL SYNOPSIS...2
Primary variable ...6
Secondary variables ...6
TABLE OF CONTENTS ...8
LIST OF ABBREVIATIONS AND DEFINITION OF TERMS...11
1. INTRODUCTION ...12
1.1 Background ...12
1.2 Rationale for conducting this NIS Primary...13
2. NIS OBJECTIVES...14
2.1 Primary objective ...14
2.2 Secondary objectives ...14
3. STUDY PLAN AND PROCEDURES ...15
3.1 Overall study design and flow chart ...16
DATA COLLECTION AT ENROLMENT AND FOLLOW-UP ...18
Enrolment procedures...18
4. SELECTION OF SUBJECT POPULATION...22
4.1 Investigators ...22
4.2 Inclusion criteria ...23
4.3 Exclusion criteria ...23
5. DISCONTINUATION OF SUBJECTS ...24
5.1 Criteria for Discontinuation ...24
5.2 Procedures for discontinuation ...24
6. THERAPEUTIC STRATEGY ...24
6.1 Therapeutic strategy of a Non-Interventional Study...24
7. STUDY CONDUCT ...24
7.1 Restrictions during the study (if applicable)...24
Date: 01 July 2015
8.1 Primary variable ...24
8.2 Variables to be collected during follow-up:...26
8.3 Patient Reported Outcomes (PRO) ...26
8.3.1 SF-36 ...26
8.3.2 Hypoglycemia Fear Survey (HFS-II)...27
8.3.3 Lifestyle score ...27
8.3.4 Healthcare avoidance due to costs ...28
8.3.5 Administration of PRO questionnaires ...28
8.4 Health Economic measurements and variables (if applicable) ...28
9. SAFETY REPORTING ...29
9.1 Definitions...29
9.1.1 Definition of Adverse Event (AE) ...29
9.1.2 Definition of Serious Adverse Event (SAE) ...29
9.1.3 Definition of Adverse Drug Reactions (ADR) ...29
9.2 Adverse Event Reporting ...29
10. ETHICAL CONDUCT OF THE NON-INTERVENTIONAL STUDY ...30
10.1 Ethics review...30
10.2 Subject Informed consent ...30
10.3 Subject data protection...31
11. STUDY MANAGEMENT BY ASTRAZENECA ...32
11.1 Monitoring, Quality Control and Archiving ...32
11.2 Training of study site personnel...33
11.3 NIS timetable and end of study...33
12. DATA MANAGEMENT...34
12.1 Collection, monitoring, processing of data and archiving ...34
12.2 Reporting and publication of data ...35
13. STATISTICAL METHODS AND SAMPLE SIZE DETERMINATION ...36
13.1 Statistical evaluation – general aspects ...36
13.2 Description of analysis sets...36
13.3 Method of statistical analysis ...36
13.4 Determination of sample size...38
Date: 01 July 2015
LIST OF TABLES
Table 1 Study Plan...22
Table 2. Precision of estimated proportions per class of products for a sample size of
200. 500 or 1000 patients (2-sided 95% Confidence Interval)...38
LIST OF FIGURES
Figure 1 Study Flow Chart...21
LIST OF APPENDICES
Date: 01 July 2015
LIST OF ABBREVIATIONS AND DEFINITION OF TERMS
The following abbreviations and special terms are used in this NIS Protocol.
Abbreviation or special term
Explanation
AE Adverse event
ADR Adverse Drug Reaction
Assessment An observation made on a variable involving a subjective judgement (assessment)
AZ AstraZeneca
CRF Case Report Form (electronic/paper)
CRO Clinical Research Organisation
ePRO Electronic Patient Reported Outcome
National Coordinator The National Coordinator is the main line of contact to coordinate the submissions and responses of the Leading Ethics Committee and of the Ethics Committees related to the other participating sites (Non-Leading Ethics Committees).
NIS Non-Interventional Study
NISA Non-Interventional Study Agreement
NISP Non-Interventional Study Protocol
NISR Non Interventional Study Report
EC Ethics Committee, synonymous to Institutional Review Board (IRB) and Independent Ethics Committee (IEC)
GCP Good Clinical Practice
ICH International Conference on Harmonisation
PI Principal Investigator responsible for the conduct of a NIS at a site
PRO Patient Reported Outcomes
Date: 01 July 2015
1.
INTRODUCTION
1.1
Background
382 million people have diabetes in 2013; by 2035 this is estimated to rise to 592 million
according to the IDF Atlas 6th edition. The number of people with type 2 diabetes is
increasing in every country. If poorly managed, diabetes can lead to serious and costly
complications.
Management of type 2 diabetes involves individualized and optimal control of factors that
cause complications, of which blood glucose is an important one of many others. Clear
guidelines for good glycaemic control have been developed recommending a glycated
haemoglobin (HbA1c) goal of <7%. Guidelines from the American Association of Clinical
Endocrinologists and the American College of Endocrinology stress the need to advance
therapy if the HbA1c goal is not met within 3 months. When anti-diabetic monotherapy cannot
adequately control blood glucose, dual anti-diabetic therapy, using a therapy with a different
mode of action, is initiated. These guidelines are based on evidence from a number of trials in
type 2 diabetes showing that tighter glycaemic control provided benefits in reducing
microvascular complications. Affordability sets a limit to the feasibility of implementation of
these guidelines.
Nevertheless, despite individualized and stricter treatment guidelines and an observed trend
towards better control, many patients are still not achieving glycaemic goals. In developing
regions, including Eastern Europe, Asia, Latin America, and Africa, the International Diabetes
Management Practices Study (IDMPS) has shown that 36% of people with type 2 diabetes
have never had their HbA1c measured, and of those with available HbA1c values, only 36%
of patients had HbA1c levels <7.0%. The Cost of Diabetes in Europe - Type 2 (CODE-2)
study evaluated glycaemic control, blood lipid levels and blood pressure, and its treatment, in
a large group of patients with type 2 diabetes in Europe. This study showed that a high
proportion of patients with risk factors for diabetes-related complications were not adequately
controlled, with just 31% of patients achieving good glycaemic control (given as
Date: 01 July 2015
study provided an update on the level of glycaemic control in Europe. The mean HbA1c value
in the CODE-2 study was 7·5% as compared to 6·9% reported in the PANORAMA study,
suggesting that glycaemic control across Europe may be improving. However, 37·4% of
patients had an HbA1c ≥7% in the PANORAMA study. Factors relating to patient
characteristics, longer duration of diabetes and more complex treatment were shown to be
strongly associated with lack of achievement of HbA1c targets.
1.2
Rationale for conducting this NIS Primary
The IDMPS, CODE-2 and PANORAMA studies were cross-sectional in their design and did
not investigate treatment patterns in terms of drug class.Little data has been captured and
published since the launch of newer classes of therapy such as DPP-IV inhibitors, GLP-1
agonists and most recently the SGLT2 inhibitors as compared to the well known established
oral hypoglyacemic agents sulphonylureas and metformin. Thus, additional studies are needed
to understand the characteristics of patients failing first line anti-diabetic therapy in the current
era, understand treatment patterns in these patients and determinants of these treatment
patterns, evaluate the effect of these strategies on improving glycaemic control and quality of
life (QoL) in the long term, and understand the unmet need for patients with type 2
diabeteschanging 1st line therapy. In this context, this study aims to provide data on real world
second and further line anti-diabetic therapy use among type 2 diabetes patients in different
geographical regions. This study will provide country-specific data and try to identify the
determinants of variability. The associations of these therapies with achieving disease control
and in preventing and controlling diabetes complications will also be documented.
In many geographical regions, data on diabetes treatments and control is scarce. Despite the
availability of some data sources there is an urgent need to conduct this study. This is because
the available data sources alone will not collect detailed data on the entire patient journey (e.g.
none will collect data on quality of life, many do not collect data on healthcare resource use).
In addition, critical data such as HbA1c, blood pressure and weight are difficult to capture
with a high level of quality, comparability and completeness. This study will collect 3 year
Date: 01 July 2015
Although available data sources will not capture the whole range of data required for
DISCOVER, established databases in certain countries (i.e. France, Germany, the Nordics and
the United Kingdom) can be further exploited to build a comprehensive data resource for
research. In other countries, electronic medical record data (such as the IMS Disease
Analyzer) and registers will be employed as a data backbone to which primarily-collected
clinical and/or patient-reported data can be linked to fill in any gaps within the patient’s
treatment journey (both prospectively and retrospectively). Where possible, DISCOVER will
also link the NIS data with existing electronic medical record sources in other countries in
order to provide further longitudinal patient data after the DISCOVER study completes. Such
a resource will allow creating a larger cohort of patients (based on the electronic medical
records) from which comparison can be made with the country-specific DISCOVER cohort.
This will be a highly innovative approach to continue to collect necessary data on type 2
diabetes patients over the long term and maximise the value of the DISCOVER program.
The present study is designed to provide real world data describing the current unmet medical
needs in patients with type 2 diabetes failing 1st line therapy and to understand the level of
disease control with current therapies, which may be very useful for both payers and
prescribers. This study serves to fill the evidence gap in the current use of second-line and
further anti-diabetic therapies, leading to a more appropriate use and long term cost
effectiveness of all anti-diabetic drugs.
2.
NIS OBJECTIVES
2.1
Primary objective
The primary objective is to describe the disease management patterns and clinical evolution
over three years in type 2 diabetes mellitus patients initiating a second line anti-diabetic
treatment, (add-on or switch), after a first line oral treatment with a monotherapy, dual or
triple therapy.
2.2
Secondary objectives
Date: 01 July 2015
To describe, overall and by patient characteristics as well as second line anti-diabetic
medication class, treatment response in terms of changes in HbA1c, body weight, blood
pressure and lipid profile from baseline and achievement of HbA1c target goals.
To describe treatment changes, that is initiation of third line or above add-on anti-diabetic
medication, initiation of insulin therapy, switching of anti-diabetic medications or dose
changes.
To describe disease progression in terms of microvascular complications, that is, incidence of
diabetic nephropathy, diabetic neuropathy, diabetic retinopathy or diabetes-related peripheral
revascularization and/or non-traumatic amputation.
To describe incidence of macrovascular complications, that is, cardiovascular death, heart
failure, myocardial infarction and stroke.
To describe hypoglycemic events and/or hospitalizations for hypoglycemia.
To describe patient reported Quality of Life.
To describe healthcare resource use.
To describe risk factors (disease, patient, treatment and setting characteristics at baseline: e.g.
age, gender, duration of diabetes, second line anti-diabetic medication class, presence of co-
morbidities, socioeconomic status, quality of care) associated with microvascular
complications, macrovascular complications, hypoglycemic events, quality of life and
healthcare resource use during follow-up.
To describe factors associated with treatment choice at baseline.
3.
STUDY PLAN AND PROCEDURES
Date: 01 July 2015
3.1
Overall study design and flow chart
This Non-Interventional Study Protocol has been subject to an internal review according to
AstraZeneca standard procedures.
This study is a multi-country, multicenter, observational, prospective, longitudinal cohort
study which will include type 2 diabetes patients initiating a second line anti-diabetic
treatment after their first line anti-diabetic therapy in a real world setting. The countries
patients will be recruited from are- Argentina, Australia, Austria, Brazil, Canada, China
(Mainland), Colombia, Costa Rica, Czech Rep, France, Germany, India, Indonesia, Italy,
Malaysia, Mexico, Netherlands, the Nordics (Denmark, Norway and Sweden) Panama,
Poland, Russia, South Korea, Spain, Taiwan and the United Kingdom.
<<The list of participating countries is not yet definitive so this list is subject to change>>.
It is estimated that approximately 9650 patients will be enrolled with each patient followed up
for 3 years. The estimated number of patients to be recruited from each country will be as
follows:
Country Number of Patients
Argentina 250
Australia 200
Austria 200
Brazil 350
Canada 500
China (Mainland) 1200
Colombia 200
Costa Rica 140
Czech Rep 200
France 500
Germany 500
India 1200
Indonesia 400
Italy 300
Malaysia 250
Date: 01 July 2015
Netherlands 200
Nordics (Denmark, Norway and Sweden) 500
Panama 60
Poland 200
Russia 500
South Korea 300
Spain 300
Taiwan 450
United Kingdom 500
Patients will undergo clinical assessments and receive standard medical care as determined by
the treating physician. Patients will not receive any experimental intervention or experimental
treatment as a consequence of their participation in the study.
To create a nationally representative sample, patients meeting the inclusion/exclusion criteria
will be consecutively recruited at study sites across geographic regions and types of sites
(office-based primary care physicians as well as endocrinologists or other types of specialists
based in hospitals with outpatient clinics), resembling the anticipated epidemiologic patterns
of T2DM in each participating country.
A Web-Based Data Capture (WBDC) system will be used for data collection and query
handling. The Investigator will ensure that data are recorded on the electronic Case Report
Form (eCRF) as specified in the study protocol and in accordance with the instructions
provided. In addition, patient-reported outcome (PRO) data will be entered into an electronic-
based database, and electronic medical records will be extracted where applicable for linkage
with the eCRF data.
Date: 01 July 2015
DATA COLLECTION AT ENROLMENT AND FOLLOW-UP
Enrolment procedures
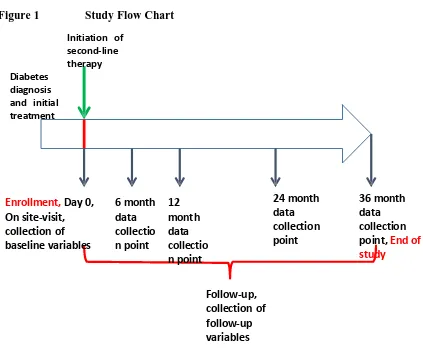
The study plan, including enrollment and follow-up visits, is outlined in Figure 1. All enrolled
patients will be followed up to the end of the study for vital status, disease evolution and
disease management.
Each patient will be invited to sign an Informed Consent Form (ICF) prior to entering the
study. Informed consent procedures are described in Section 10.2. Subjects who meet
inclusion/exclusion criteria will be enrolled consecutively at a study site. In addition, in order
to compensate for the different overall recruitment time periods between countries,
retrospective identification of subjects will be allowed. All subjects who meet the study
inclusion/exclusion criteria and initiated their 2nd line antidiabetic treatment on December
31st, 2014, or afterwards can be invited to participate in the study.
Where local regulations allow, investigators will record the minimum information of all
subjects who meet inclusion criteria regardless of whether they agree to participate or not onto
a screening log (i.e., gender, age and the reason of non-inclusion) to assess potential selective
enrollment of subjects.
In addition, consenting eligible subjects will be requested to complete a Contact Order Form,
in which the subject will confirm his/her agreement to be contacted by the study site if the
subject does not attend routine visits. Subjects will be requested to complete their contact
details on this form, as well as those of the person who the study site could contact in case the
subject remains unreachable after several attempts (to check vital status) including preferably
a person living in the same household. The contact details which will be supplied by the
subject on the Contact Order Form will be kept solely by the study site and study sponsor and
CRO personnel will not be privy to this information. At the end of the study, this information
(on paper) will be deleted/destroyed by the study site.
Patients will also be asked to provide consent to link the information captured in the eCRF
Date: 01 July 2015
laboratory values that may be captured in electronic records and linked in the future to
enhance the data capture for each patient, where technically and ethically feasible.
A unique study identification code for the subject will be assigned by the site created through
the eCRF, which which will also be assigned to the corresponding electronic medical record
for linkage (where applicable). In the case of linking the eCRF records to other patient-level
health information, secondary codes from each additional source of health information will
also be matched to the unique primary study code generated for each subject. Patient
information will be linked together using a set of characteristics (such as name, zip code, etc.)
by a third party, who will securely hold the key to the patient linkage (in accordance with
local privacy laws) and will generate a unique code for the full linked set of health
information to maintain patient anonymity and data integration at all times during research.
1.-Enrollment/Baseline data collection (Visit 1)
Potentially eligible patients approached by investigators meeting the inclusion and exclusion
criteria and following their signed informed consent will be reviewed. Initial data collection
will be performed by the investigator. As collection of data might not be possible during a
routine clinical visit (due to time constraints), the investigator is allowed to arrange a visit
with the patient within the 2-weeks following prescription of the 2nd line diabetic medication,
to collect all the necessary information. This will include demographic data, vital signs and
available results of lab tests, medical history of T2DM, presence of co-morbidities and co-
medication use, previous (1st line) diabetic medication and reason for change, 2nd line diabetic
medication (by class), HbA1c target and patient-reported Quality of Life. Patients identified
retrospectively, meeting the inclusion and exclusion criteria and initiating their 2nd line
antidiabetic treatment on December 31st, 2014 or afterwards can also be included. The
investigator will collect “baseline” data from these patients at the time of inclusion (i.e., after
informed consent signature).“Baseline” data will consist of the information corresponding to
the moment when the 2nd line antidiabetic treatment was initiated or previous to it, e.g.,
demographic data, vital signs and available results of lab tests, medical history of T2DM,
Date: 01 July 2015
reason for change, 2nd line diabetic medication (by class) and HbA1c target. Patients identified
retrospectively will not complete the patient-reported Quality of Life questionnaires at baseline.
2.- Follow up data collection (Visit 2, 3, 4, 5 are on-site visits at month of 6, 12, 24, 36)
The follow-up period for each patient is from enrollment up to 36 months.
During this period, patients will attend the study site for routine visits. Data will be captured
from patient records at 6 month, 12 month, 24 month, and 36 month time points with a ±2
month buffer period. The investigator may contact the patient via telephone to obtain
necessary information.
During the follow-up interviews with the patients, the following data will be collected:
physiological parameters and lab tests, changes in diabetic medications and their reasons
(including HbA1c at the time of change), incidence of microvascular complications
(nephropathy, retinopathy, neuropathy and amputation), incidence of macrovascular events,
incidence of major hypoglycemic events and occurrence of minor hypoglycemic events,
changes in co-morbidities and co-medications, patient-reported quality of life and healthcare
resource use. For patients identified retrospectively, any information generated after the 2nd
line antidiabetic treatment was initiated will be considered as follow-up information and will
be collected at the time of patient inclusion (after informed consent signature) or in
subsequent follow-up data collection time points. Patients identified retrospectively will
complete the patient-reported quality of life questionnaires during follow-up similarly to all
other study participants (i.e., only the baseline questionnaires will be missing for these
patients).
In the countries where an electronic medical record backbone will be used as the basis for data
capture, an additional questionnaire will be provided to physicians to complete information that
Date: 01 July 2015
missing values). This additional data capture will be linked back to the electronic medical
record to create a full data resource for each patient.
Figure 1 Study Flow Chart
Diabetes diagnosis and initial treatment
Initiation of second-line therapy
Enrollment, Day 0, On site-visit, collection of baseline variables
6 month data collectio n point
12 month data collectio n point
24 month data collection point
36 month data collection
point, End of
study
Date: 01 July 2015
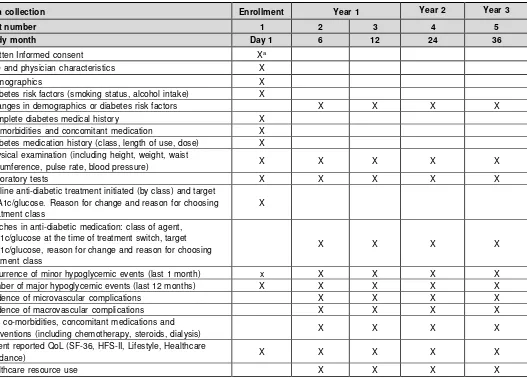
Table 1 Study Plan
Data collection Enrollment Year 1 Year 2 Year 3
Visit number 1 2 3 4 5
Study month Day 1 6 12 24 36
Written Informed consent Xa
Site and physician characteristics X
Demographics X
Diabetes risk factors (smoking status, alcohol intake) X
Changes in demographics or diabetes risk factors X X X X
Complete diabetes medical history X
Co-morbidities and concomitant medication X
Diabetes medication history (class, length of use, dose) X
Physical examination (including height, weight, waist
circumference, pulse rate, blood pressure) X X X X X
Laboratory tests X X X X X
2nd line anti-diabetic treatment initiated (by class) and target
HbA1c/glucose. Reason for change and reason for choosing treatment class
X
Switches in anti-diabetic medication: class of agent, HbA1c/glucose at the time of treatment switch, target HbA1c/glucose, reason for change and reason for choosing treatment class
X X X X
Occurrence of minor hypoglycemic events (last 1 month) x X X X X
Number of major hypoglycemic events (last 12 months) X X X X X
Incidence of microvascular complications X X X X
Incidence of macrovascular complications X X X X
New co-morbidities, concomitant medications and
interventions (including chemotherapy, steroids, dialysis) X X X X
Patient reported QoL (SF-36, HFS-II, Lifestyle, Healthcare
avoidance) X X X X X
Healthcare resource use X X X X
a Informed consent must be obtained before data collection is started
4. SELECTION OF SUBJECT POPULATION
4.1 Investigators
The selection of sites and investigators will be made with an aim to achieve representativeness
of the way T2DM is managed in each participating country. The National Coordinator of each
country will provide advice regarding the proportion of the different types of centres (primary
Date: 01 July 2015
different types of specialists) involved in T2DM management. They will also provide an
insight on the national situation regarding anti-diabetic medication prescription
(reimbursement, public or private insurance). All this information will be used to select sites
and investigators for the study resembling as closely as possible that situation. Sites will also
be evaluated for existence and use of electronic medical records as part of feasibility
determination for a future data extraction network.
4.2
Inclusion criteria
The subject population that will be observed in the NIS, must fulfil all of the following
criteria:
1. Provision of subject informed consent
2. Female or male aged 18 years and over
3. Diagnosed with type 2 diabetes mellitus
4. Initiating a second line anti-diabetic therapy (add-on or switching) after a first line
oral treatment with a monotherapy, dual or triple therapy.
The prescription of the medicinal product is clearly separated from the decision to include the
subject in the NIS.
4.3
Exclusion criteria
Patients will not be eligible to participate if any of the following exclusion criteria are present:
1. Diagnosis of type 1 diabetes mellitus
2. Patient is pregnant
3. Patients initiating a dual therapy after having previously received two different lines of
monotherapy before (e.g.: Metformin → SU → SU+Add-on)
4. Current treatment with chemotherapy, oral or iv steroids
5. Patient is on dialysis or has had a renal transplant
6. The patient is taking insulin or an injectable agent as first line treatment.
Date: 01 July 2015
9. Presence of any condition/circumstance which in the opinion of the investigator could
significantly limit the complete follow up of the patient for 3 years (e.g. life-
threatening co-morbidities, tourist, non-native speaker or does not understand the
local language where interpreter services are not reliably available, psychiatric
disturbances, dementia, alcohol or drug abuse).
10. Not willing to sign the informed consent.
5. DISCONTINUATION OF SUBJECTS
5.1 Criteria for Discontinuation
As a patient’s participation is voluntary, patients are free to discontinue their participation in
this study at any time and without prejudice to his/her subsequent medical treatment. Patients
who withdraw prematurely from this study will not be replaced.
5.2 Procedures for discontinuation
Subjects who discontinue should be asked about the reason(s) for their discontinuation, and a
follow up call will be made at the time of withdrawal for final data collection, if possible.
6. THERAPEUTIC STRATEGY
6.1 Therapeutic strategy of a Non-Interventional Study
The assignment of a subject to a particular therapeutic strategy is not decided in advance by a protocol but falls within current practice.
7. STUDY CONDUCT
7.1
Restrictions during the study
(if applicable)There are no specific restrictions in this study.
8.
MEASUREMENTS OF STUDY VARIABLES AND
DEFINITIONS OF OUTCOME VARIABLES
8.1 Primary variable
Date: 01 July 2015
o Centre type (primary care centre, University hospital, General hospital,
Specialized diabetes centre).
o Geographical setting (urban, rural)
o Centre funding (public/private/mixed)
• Physician speciality (GP, Endocrinologist, Internist, Cardiologist, other specialists)
• Patient demographics (gender, date of birth, self reported ethnic origin, living
arrangement status -living alone, not living alone, education level (basic/low,
medium/high), employment status (yes or no)).
• Co-morbidities and co-medications.
• Medical history of T2DM
o Date of diagnosis
o History of diabetes treatment (class, duration of treatment, dose)
o Evidence of microvascular complications (nephropathy, retinopathy,
neuropathy and amputation)
o Number of any major hypoglycemic events and/or hospitalization for
hypoglycemic events in the past 12 months
o Occurrence of minor hypoglycemic events in the last 1 month
o Patient received diabetes education
o Risk factors (alcohol intake, smoking status)
• Vital signs (weight, height, BMI, waist circumference, seated blood pressure (SBP and
DBP), pulse rate).
• Laboratory tests (most recent results available within the last 3 months):
o Blood test data: HbA1c (at the time of 2nd line treatment initiation), glucose
(fasting, post-prandial or casual), aspartate aminotransferase (AST), alanine
aminotransferase (ALT), Gamma glutamyl transpeptidase (γ GTP),
creatinine,albumin, hematocrit, white cell blood count, platelets, hemoglobin,
total cholesterol, LDL-C, HDL-C, triglycerides, uric acid.
o Urinary test data: urine protein, urine albumin, urinary creatinine.
• 2nd line anti-diabetic treatment initiated
Date: 01 July 2015
o HbA1c/glucose target for patient
o Reasons for initiating a 2nd line therapy.
o Reasons for choosing second line treatment
• Patient-reported outcomes: Quality of Life (SF-36, HFS-II, lifestyle and healthcare
avoidance due to cost questionnaires), except for patients identified retrospectively
8.2
Variables to be collected during follow-up:
• Changes in vital signs
• Changes in diabetes treatments (date of change, drug class changed to, change made-
(add-on, discontinuation or switching), reason for change, reason for choosing drug
class, dose, HbA1c/glucose target for patient).
• Number of major hypoglycemic events since last visit.
• Occurrence of minor hypoglycemic events in the last 1 month.
• Microvascular complications (nephropathy, retinopathy, neuropathy and amputation).
• Macrovascular complications (myocardial infarction, heart failure and stroke).
• Changes in co-morbidities and co-medications.
• All-cause death.
• Patient reported outcomes: Quality of Life: SF-36, HFS-II, lifestyle and healthcare
avoidance due to cost.
• Healthcare resource use (hospitalisations -emergency and elective-, procedures, use of
glucose monitoring devices)
8.3
Patient Reported Outcomes (PRO)
The specific PROs to be used in each country will depend on the availability of versions in the
official language/s.
8.3.1 SF-36
The SF-36 is a multipurpose short form survey with 36 questions, measuring 8 health
domains: physical functioning, role physical, bodily pain, general health, vitality, social
functioning, role emotional, and mental health. Physical functioning covers limitations in
daily life due to health problems. The role physical scale measures role limitations due to
Date: 01 July 2015
with usual roles. The general health scale measures individual perceptions of general health.
The vitality scale assesses energy levels and fatigue. The social functioning scale measures the
extent to which ill health interferes with social activities. The role emotional scale assesses
role limitations due to emotional problems, and the mental health scale measures
psychological distress. The SF-36 can also be used to derive 2 aggregate summary measures:
the Physical and Mental Health Composite Scores (PCS & MCS). Summary scores are
calculated by summing factor-weighted scores across all 8 subscales, with country specific
factor weights. The scores range from 0 to 100, where a zero score indicates the lowest level
of health measured by the scales and 100 indicates the highest level of health. The SF-36 is
available in 2 forms: a standard form, which uses a 4-week recall period, and an acute form,
which uses a 1-week recall. The standard 4-week recall form is appropriate when at least 4
weeks will pass between re-administration of the instrument.
8.3.2 Hypoglycemia Fear Survey (HFS-II)
The revised Hypoglycemia Fear Survey (HFS-II) is used to measure behaviours and worries
related to fear of hypoglycemia in adults with diabetes. The HFS-II is composed of two
subscales, the Behaviour (HFS-B) and Worry (HFS-W). HFS-B items describe behaviours in
which patients may engage to avoid hypoglycemic episodes and/or their negative
consequences (e.g., keeping blood glucose [BG] levels high, making sure other people are
around, and limiting exercise or physical activity). HFS-W items describe specific concerns
that patients may have about their hypoglycemic episodes (e.g., being alone, episodes
occurring during sleep, or having an accident). The HFS-II is a 33-item questionnaire with
two subscales that measure 1) behaviours to avoid hypoglycaemia and its negative
consequences and 2) worries about hypoglycaemia and its negative consequences. Responses
are made on a 5-point Likert scale where 0 = Never and 4 = Always. There is a score range of
0 - 132 with higher scores indicating increased fear of hypoglycaemia.
8.3.3 Lifestyle score
We will assess patient lifestyle using a previously published questionnaire. All lifestyle
Date: 01 July 2015
factors covered are: smoking habits, alcohol intake, physical activity, dietary intake of fish,
processed meats, fruits and vegetables.The score from a maximum of seven possible
healthy lifestyle factors are divided into four lifestyle groups: unhealthy (none, one, or
two healthy lifestyle factors; 0–2), intermediate (three healthy lifestyle factors; 3),
healthy (four or five healthy lifestyle factors; 4–5), and very healthy (six or seven
healthy lifestyle factors; 6–7).
8.3.4 Healthcare avoidance due to costs
This will be evaluated using a questionnaire consisting of two questions assessing whether or
not a patient has avoided a) healthcare and/or b) medication due to cost.
8.3.5 Administration of PRO questionnaires
The PRO questionnaires will be:
z Self-administered during the site visit at enrollment and follow-up phase
z Through telephone interview during the follow-up phase (if applicable)
Patients will be asked to complete the questionnaires during their site visits. See Table1 for
detailed frequency of each questionnaire.
8.4
Health Economic measurements and variables (if applicable)
Resource use such as hospitalizations, investigations, procedures /interventions,
visits to GP, specialist, emergency visits, investigations, procedures will be collected.
Healthcare resource use will be collected and analyzed on all patients in the study.
Direct healthcare costs will be estimated by multiplying resource use with country specific
Date: 01 July 2015
9.
SAFETY REPORTING
9.1
Definitions
9.1.1 Definition of Adverse Event (AE)
An adverse event is the development of an undesirable medical condition or the deterioration
of a pre-existing medical condition following or during exposure to a pharmaceutical product,
whether or not considered causally related to the product. An undesirable medical condition
can be symptoms (eg, nausea, chest pain), signs (eg, tachycardia, enlarged liver) or the
abnormal results of an investigation (eg, laboratory findings, electrocardiogram). In clinical
studies, an AE can include an undesirable medical condition occurring at any time, including
run-in or washout periods, even if no study treatment has been administered
9.1.2 Definition of Serious Adverse Event (SAE)
A serious adverse event is an AE occurring during any study phase and fulfils one or more of the following criteria:
• results in death
• is immediately life-threatening
• requires in-patient hospitalisation or prolongation of existing hospitalisation
• results in persistent or significant disability or incapacity
• is a congenital abnormality or birth defect
• is an important medical event that may jeopardise the subject or may require
medical intervention to prevent one of the outcomes listed above
9.1.3 Definition of Adverse Drug Reactions (ADR)
An ADR is the development of an undesirable medical condition or the deterioration of a pre-
existing medical condition following or during exposure to a medicinal product, suspected to
be causally related to the product
9.2
Adverse Event Reporting
As this is a non-interventional study where patient selection is not based on the treatment
Date: 01 July 2015
reactions (ADRs) observed in subjects participating in this study, should be reported to Health
Authorities as stated in local regulations, and/or, if the investigator considers it appropriate.
10.
ETHICAL CONDUCT OF THE NON-INTERVENTIONAL
STUDY
The Non-Interventional Study will be performed in accordance with ethical principles that are
consistent with the Declaration of Helsinki, ICH GCPs and the applicable legislation on Non-
Interventional Studies.
The Investigator will perform the NIS in accordance with the regulations and guidelines
governing medical practice and ethics in the country of the NIS and in accordance with
currently acceptable techniques and know-how.
10.1
Ethics review
The final protocol of the Non-Interventional Study, including the final version of the Subject
Informed Consent Form, must be approved or given a favourable opinion in writing by the
Ethics Committee.
The Ethics Committee must also approve any amendment to the protocol and all advertising
used to recruit subjects for the study, according to local regulations.
10.2
Subject Informed consent
The Investigator at each site will ensure that the subject is given full and adequate oral and
written information about the nature, purpose, possible risk and benefit of the NIS. Subjects
must also be notified that they are free to discontinue from the NIS at any time. The subjects
should be given the opportunity to ask questions and allowed time to consider the information
provided.
The signed and dated subject informed consent must be obtained before any specific
procedure for the NIS is performed, including:
• Interview with the investigator
Date: 01 July 2015
• CRFs completion.
• Use of other possible sources of health information, such as electronic health record
or laboratory values that may be captured in electronic records and linked in the
future to enhance the data capture for each patient, where technically and ethically
feasible.
The Investigator must store the original, signed Subject Informed Consent Form. A copy of
the signed Subject Informed Consent Form must be given to the subject.
Subjects will also be asked to complete and sign a Contact Order Form, with their own contact
details as well as those of a relative or close person (preferably a person living in the same
household), and authorization to be contacted by the study site if they do not attend routine
visits or remain unreachable after several attempts (to check vital status).
10.3
Subject data protection
The Subject Informed Consent Form will incorporate wording that complies with relevant
data protection and privacy legislation. Pursuant to this wording, subjects will authorise the
collection, use and disclosure of their personal data by the Investigator and by those persons
who need that information for the purposes of the NIS.
The Subject Informed Consent Form will explain that NIS data will be stored in a computer
database, maintaining confidentiality in accordance with the local law for Data Protection.
The Subject Informed Consent Form will also explain that for quality check purposes, a
monitor of AZ or a monitor of company representing AZ, will require direct access to the
signed subject informed consent forms. In case source data verification will be planned as
quality check, the Subject Informed Consent Form will explain that for data verification
purposes, monitor of AZ or a monitor of company representing AZ may require direct access
to source documents that are part of the hospital or practice records relevant to the Non-
Date: 01 July 2015
11.
STUDY MANAGEMENT BY ASTRAZENECA
11.1
Monitoring, Quality Control and Archiving
Before the first subject is recruited into the study, the local representative of AZ or of a
company representing AZ will:
• Establish the adequacy of the facilities and the investigator’s capability to
appropriately select the sample
• Discuss with the investigator(s) (and other personnel involved with the study) their
responsibilities with regards to protocol compliance, and the responsibilities of
AstraZeneca or its representatives. This will be documented in a NIS Primary
Agreement between AstraZeneca/delegate and the investigator.
During the study the local representative of AZ or of a company representing AZ can
implement different activities to assure compliance with AZ standards of quality. These
activities could include but are not limited to:
Contacts with the sites to:
• Provide information and support to the investigator(s)
• Confirm that the research team is complying with the protocol and that data are
being accurately recorded in the case report forms (CRFs)
• Ensure that the subject informed consent forms are signed and stored at the
investigator’s site
• Ensure that the CRFs and PROs are completed properly and with adequate quality.
• Work with the site on the extraction of electronic medical records (EMRs)
Monitoring activities for:
• Checking a sample of ICFs
• Checking that subjects exist in medical records (a sample)
• Checking a few (4 or 5) key variables in the CRF against the original medical
Date: 01 July 2015
• In addition, methods for validating linkage of the CRF and PRO with electronic
medical record additional health data resources (where applicable) will be used.
Different signals (eg, high rejection rate in a site) should be used as potential identification of
low protocol compliance by investigators.
If these, or any other signal occurs or if the local coordinator is suspicious of a potential non-
optimal level of protocol compliance by the site investigator, specific measures should be
adopted to evaluate the situation, identify the issue and implement specific action plans to
correct the situation.
11.2
Training of study site personnel
The Principal Investigator will ensure that appropriate training relevant to the NIS is given to
investigational staff, and that any new information relevant to the performance of this NIS is
forwarded to the staff involved.
11.3
NIS timetable and end of study
Before the first subject is enrolled in the NIS and any NIS related procedures are undertaken
the following should be fulfilled
• Written approval of the NIS by the Ethics Committee and/or Regulatory
Authorities, according to local regulations
• Proper agreements between AstraZeneca and the Investigator/Institution is signed
• Proper agreements are in place with the EMR providers if electronic medical
records are extracted
The planned timetable for the NIS is estimated to be as follows:
• Estimated first subject in: Q4 2014
• Estimated last subject in: Q4 2015
• Estimated last subject last visit: Q4 2018
Date: 01 July 2015
Should AstraZeneca decide to discontinue the study prior to what was established in this
protocol, the investigator, and relevant authorities should receive written notice describing the
reasons why the study was terminated at an earlier date. The investigator will immediately
notify the subjects taking part in the study; they will continue to receive their treatment
according to usual clinical practice.
12.
DATA MANAGEMENT
12.1
Collection, monitoring, processing of data and archiving
Data management will be performed by a vendor selected by AstraZeneca, which will be
responsible for the set up of the study electronic CRF.
Data will be entered in the WBDC system at the Investigator’s site. The Investigator will be
responsible for entering data into the WBDC system and according to the Investigator
Instructions Manual. The Investigator Instructions Manual will also provide the site with data
entry instructions.
Data entered in the WBDC system will be immediately saved to a central database and
changes tracked to provide an audit trail. When data have been entered, reviewed and edited,
the Investigator will be notified to sign the e-CRF electronically as per the agreed project
process and data will be locked to prevent further editing. A copy of the e-CRF will be
archived at the Investigator’s site.
Real-time reports will be generated to validate data during the study.
All CRFs will be checked to ensure that they are properly completed with adequate quality.
This check does not necessarily need to be performed at the study sites.
When all data have been recorded, checked, signed and locked, AstraZeneca will declare a
clean file.
A copy of the eCRF will be archived at the Investigator’s site for a period agreed in the site
contract.
The CRF will be linked at a patient level to the records generated from electronic medical
records and/or registries in accordance with local patient privacy guidelines if technically
Date: 01 July 2015
PRO data will also be entered into a web-based validated software system. All data related to
patient reported outcomes will be captured pseudonymously using a patient identification
number. The patient identification number allows matching the patient reported data with the
patient data collected by the physicians.
12.2
Reporting and publication of data
• AstraZeneca will prepare a Non-Interventional Study Report within 12 months after
completion of the last subject.
• The AstraZeneca Medical VP or delegate will review and sign off the study report.
• AstraZeneca will communicate the study results to all participating investigators.
• AstraZeneca is obliged to analyze and report all study data as described in the
protocol.
In accordance with the Declaration of Helsinki, both authors and publishers have ethical
obligations. In publication of the results of the NIS, the authors are obliged to preserve the
accuracy of the results. Negative as well as positive results should be published or otherwise
publicly available. AstraZeneca endeavours to publish the results of NIS and is committed to
ensure that the data are reported in a responsible and coherent manner.
AstraZeneca seeks to ensure that publications in biomedical journals follow the guidelines
established by the International Committee of Medical Journal Editors (ICMJE) and published
in its Uniform Requirements of Manuscripts Submitted to Biomedical Journals.
AstraZeneca is committed to ensuring that authorship for all publications should comply with
the criteria defined by the ICMJE. These state that: "Each author should have participated
sufficiently in the work to take public responsibility for the content."
AstraZeneca believes that participation solely in the collection of data or drafting the
manuscript does not justify authorship. These conditions apply equally to external
investigators and to AstraZeneca employees.
Date: 01 July 2015
Publication of data subsets from individual institutions participating in multicentre studies
should not precede the primary manuscript, and when developed should always reference the
primary publication of the entire study.
13.
STATISTICAL METHODS AND SAMPLE SIZE
DETERMINATION
13.1
Statistical evaluation
–
general aspects
A Non-Interventional Study is a study in which epidemiological methods including other
methods that can be used to analyse human population health data.
All statistical analysis will be performed by the vendor selected by AstraZeneca by means of
the SAS statistical software system (SAS Institute, Cary, NC).
A comprehensive Statistical Analysis Plan will be prepared before database lock.
13.2
Description of analysis sets
The study population for analysis will consist of all patients enrolled into the study.
Patients identified as potentially eligible and non-consenting, will be described anonymously
using data provided by the physician.
For descriptive baseline analyses, all enrolled patients for who the relevant data are available
will be included. For the follow up period all enrolled patients with at least one follow up
contact recorded will be included in descriptive analyses.
Time to event analysis will use the study population of all enrolled patients.
The number of subjects with any follow-up data will be presented, and the reasons for lack of
contact will be summarised.
13.3
Method of statistical analysis
Date: 01 July 2015
Demographic variables, patient characteristics and treatment patterns will be summarized
using descriptive statistics.
Descriptive statistics will include n, mean, median, standard deviation, minimum and
maximum for continuous variables and frequency for categorical variables. Two-sided 95%
confidence intervals can be obtained if considered relevant.
Identification of predictors of treatment choice using baseline characteristics will be attempted
using multivariate regression.
Changes in HbA1c, blood glucose, lipid profile, body weight and blood pressure will be
summarised with descriptive statistics. Stratifications will be made by anti-diabetic class at
baseline and regression models will be used to see if anti-diabetic class at baseline is a
predictor of these outcomes.
Proportions of patients switching and/or adding treatments and/or changing doses of
treatments will be summarised with descriptive statistics.
Time to event survival analyses (Switching of the 2nd line drug, initiation of insulin therapy
and initiation of the 3rd or above anti-diabetic therapy) will be performed. Multivariate Cox
models will be applied to analyze time to event data in order to assess the association of
treatment class at baseline with clinical outcome variables.
Proportions of patients with diabetic nephropathy; diabetic neuropathy; diabetic retinopathy;
peripheral revascularization and/or non-traumatic amputation; major hypoglycemia will be
summarised descriptively. Stratifications will be made by anti-diabetic therapy class at baseline
and regression models will be used to see if anti-diabetic therapy class at baseline is a predictor
of these outcomes.
Healthcare resource use and quality of life results will be summarized descriptively.
Stratifications will be made by anti-diabetic therapy class at baseline and regression models
Date: 01 July 2015
Interim analysis might be performed at baseline after the last subject is recruited and then 1
and 2 years after last subject in.
13.4 Determination of sample size
In the DISCOVER study, the sample size will ensure that the descriptive data mandated by the
primary objective in relation to disease, patient and management patterns characteristics are
sufficiently precise and meaningful at a country level.
With a sample size of at least 200 patients for the different participating countries (for
example, the Central American Carribean Region countries will be analysed together), the
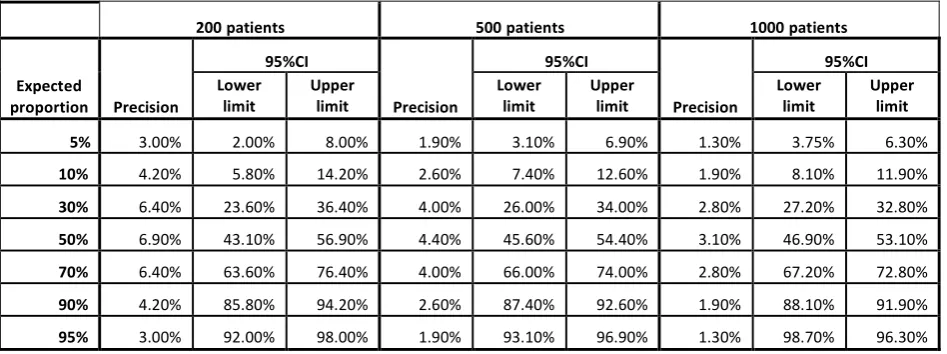
[image:40.596.80.554.350.527.2]estimated precision for any given characteristic proportion is shown in the Table below.
Table 2. Precision of estimated proportions per class of products for a sample size of 200. 500 or 1000 patients (2-sided 95% Confidence Interval)
200 patients 500 patients 1000 patients
Expected proportion Precision 95%CI Precision 95%CI Precision 95%CI Lower limit Upper limit Lower limit Upper limit Lower limit Upper limit
5% 3.00% 2.00% 8.00% 1.90% 3.10% 6.90% 1.30% 3.75% 6.30%
10% 4.20% 5.80% 14.20% 2.60% 7.40% 12.60% 1.90% 8.10% 11.90%
30% 6.40% 23.60% 36.40% 4.00% 26.00% 34.00% 2.80% 27.20% 32.80%
50% 6.90% 43.10% 56.90% 4.40% 45.60% 54.40% 3.10% 46.90% 53.10%
70% 6.40% 63.60% 76.40% 4.00% 66.00% 74.00% 2.80% 67.20% 72.80%
90% 4.20% 85.80% 94.20% 2.60% 87.40% 92.60% 1.90% 88.10% 91.90%
95% 3.00% 92.00% 98.00% 1.90% 93.10% 96.90% 1.30% 98.70% 96.30%
This means that regarding any qualitative variable at a frequency of 5% to 95%, and for
countries with a sample size of 200 patients, the precision would range from 3.0% to 6.9%
which is scientifically acceptable. For countries with 500 patients, the precision would range
from 1.9% to 4.4% at a 95% confidence limit and for countries with a sample size of 1000
patients, the precision would be even better, ranging from 1.3% to 3.1.
Analysis of stratified data, however, will in some cases have to be done only at a global level,
and not at a country level, to ensure sufficient precision and reliability of the data. A typical
example would be analyses of patients given a specific compound class. Recent Ipsos
Date: 01 July 2015
compound classes are given to 1%-10% patients as a second line (either as monotherapy, dual
therapy or triple therapy). If we take as an example a class of compounds given to 1.5% of the
population, this would represent 8 patients treated with that class in a country including 500
patients, or 15 patients in a country including 1000 patients. These numbers are too small to
allow sufficient precision for any type of analyses, thus precluding by-country analyses.
However, if the stratified analyses by class of agent are done with the overall study population
(9650 patients), there would be 201 patients treated with that class, leading to scientifically
acceptable precisions.
14. LIST OF REFERENCES
1. International Diabetes Federation. IDF Diabetes Atlas, 6th edn. Brussels, Belgium: International Diabetes Federation, 2013.
2. Chan JC, Gagliardino JJ, Baik SH, Chantelot JM, Ferreira SR, Hancu N, Ilkova H, Ramachandran A, Aschner P; IDMPS Investigators. Multifaceted determinants for achieving glycemic control: the International Diabetes Management Practice Study (IDMPS). Diabetes Care. 2009 Feb;32(2):227-33. doi: 10.2337/dc08-0435. Epub 2008 Nov 25. PubMed PMID: 19033410; PubMed Central PMCID: PMC2628684.
3. de Pablos-Velasco P, Parhofer KG, Bradley C, Eschwège E, Gönder-Frederick L, Maheux P, Wood I, Simon D. Current level of glycaemic control and its associated factors in patients with type 2 diabetes across Europe: data from the PANORAMA study. Clin Endocrinol (Oxf). 2014 Jan;80(1):47-56. doi: 10.1111/cen.12119. Epub 2013 May 6. PubMed PMID: 23194193.
4. Liebl A, Mata M, Eschwège E; CODE-2 Advisory Board. Evaluation of risk factors for development of complications in Type II diabetes in Europe. Diabetologia. 2002 Jul;45(7):S23-8. Epub 2002 Jun 19. PubMed PMID: 12136408.
5. Carlsson AC, Wändell PE, Gigante B, Leander K, Hellenius ML, de Faire U. Seven modifiable lifestyle factors predict reduced risk for ischemic cardiovascular