BAB 2
TINJAUAN PUSTAKA
2.1. THALASSEMIA
2.1.1. Defenisi
Istilah talasemia berasal dari kata Yunani yaitu Thalassa (laut) dan Haema (darah) yang mengacu pada adanya gangguan sintesis dari rantai globin (rantai α dan rantai β) yang merupakan subunit dari hemoglobin Hb A (α2; β2) ¹⁹˒²º. Gen untuk sintesis rantai globin terletak di kromosom 11 (β) dan 16 (α)²¹. Sindrom Thalasemia diklasifikasikan berdasarkan adanya gangguan dari rantai globin, α atau β¹⁸. Thalassemia adalah kelainan herediter yang ditandai dengan tidak adekuatnya sintesis dari satu atau lebih rantai dari globin³‚¹⁴.
2.1.2. Struktur dan Sintesis Hemoglobin
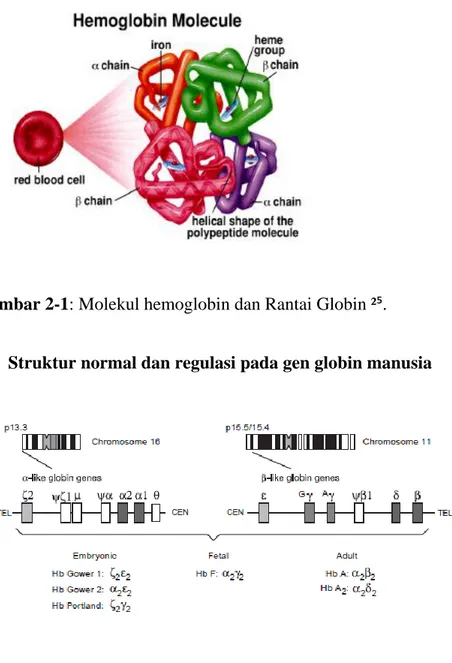
Hemoglobin merupakan pigmen yang terdapat didalam eritrosit yang terdiri dari heme dan globin dan memiliki berat molekul 64-64.4 kDa. Molekul hemoglobin yang terkandung dalam sel-sel darah merah sangat penting untuk kehidupan manusia. Heme sangat penting untuk transportasi oksigen sedangkan globin berfungsi untuk melindungi heme dari oksidasi. Struktur molekul hemoglobin menghasilkan lingkungan internal hidrofobik yang melindungi besi pada heme dari air, dan juga dari oksidasi²².
Hemoglobin berbentuk heterotetramer yang terdiri dari dua pasang rantai polipeptida yang berkaitan dengan gen α-globin (α like globins) dan dua pasang rantai polipeptida yang berhubungan dengan gen β-globin (β-like globins). Rantai Globin polipeptida akan mengikat heme, yang nantinya hemoglobin di eritrosit berfungsi untuk mengangkut oksigen dan sebagai transportasi oksigen dari paru-paru ke jaringan²³˒²⁴
Gambar 2-1: Molekul hemoglobin dan Rantai Globin ²⁵.
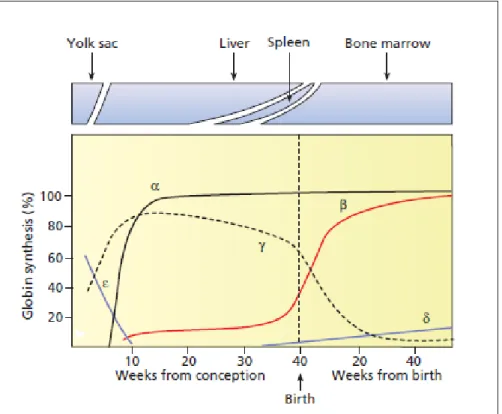
Struktur normal dan regulasi pada gen globin manusia
Gambar 2-2: Struktur rantai gen globin pada kromosom 16 dan 11 pada fase embrional ,
fetus dan dewasa ²³ (sumber: Disorder Hemoglobin).
Lokus gen globin pada β-globin terletak pada kromosom 11, dan lokus gen α-globin terletak pada kromosom 16²¹. Urutan aktivasi dimulai dari masa embrional sampai dewasa dari gen δ ke α dari gen Ɛ ke ϒᴳ,ϒᴬ,δ dan β. Maka hemoglobin utama pada masa embrional adalah Hb Gower 1 (δ₂Ɛ₂), Hb Gower 2 (α₂Ɛ₂), dan Hb Portland (δ₂ϒ₂). Pada masa janin sampai perinatal adalah HbF(α₂ϒ₂), dan pada anak yang berumur lebih dari 1 tahun sampai dewasa normal terdiri dari HbA (α₂β₂) dan HbA2 (α₂δ₂)¹⁴˒²³˒²⁶. Pada 6 bulan pertama perkembangan janin kehidupan neonatal, terjadi pola yang kompleks dari ekspresi gen globin yang disebut hemoglobin switch²⁴. Pada awal kehidupan embrional sampai delapan minggu
sintesis rantai globin akan disintesis yolk sac dan hati yaitu rantai δ yang berkombinasi dengan rantai Ɛ akan membentuk Hb Gower 1, Hb Gower 2 dan Hb Portland. Ekspresi yang singkat dari gen globin pada masa embrio, maka pada akhir kehamilan akan dibentuk hemoglobin utama pada janin yaitu Hemoglobin F (α2γ2) dan organ yang terlibat dalam sintesis rantai globin tersebut adalah hati, limpa dan sumsum tulang. kemudian akan digantikan oleh rantai β-globin dewasa yaitu hemoglobin A (α2β2), hemoglobin A2 (α₂δ₂) dan Hemoglobin F (α₂ϒ₂) yang kadarnya <0,5%¹²˒²³˒²⁷.
2.1.3 Klasifikasi
Klasifikasi dari Thalassemia berdasarkan jenis subunit globin yang mengalami defek, dan secara garis besar terdiri dari:
2.1.3.1 Thalassemia-α
Hilangnya produksi gen α (α⁰) atau berkurangnya produksi dari gen α (α⁺), disebabkan oleh mutasi gen globin α baik berupa delesi gen maupun non-delesi (mutasi titik). Suatu studi molekul yang menggunakan teknik hibrid telah mengidentifikasi hilangnya fungsi gen α yang terkait delesi atau nondelesi dari mutasi gen menyebabkan berkurangnya fungsi gen sehingga menyebabkan mutasi pada kodon yang bertanggung jawab terjadinya syndrom α-thalassemia ¹⁸˒²⁸. Pada α-thalassemia pembagiannya tergantung pada jenis mutasi gen-α yang mengalami kerusakan. Secara klinis thalassemia-α dapat terbagi menjadi 4 kelompok:
1. Silent thalassemia-α (-α/αα).
Delesi 1 rantai α . Selalunya disebut thalassemia-α⁺. Pada keadaan ini tidak terjadi kelainan hematologi . Kelainan ini ditemukan sekitar 15-20% dari populasi keturunan Afrika.
2. Carrier thalassemia-α (--/αα atau –α/-α)
Delesi pada 2 gen-α. Disebut juga thalassemia-α minor. Dijumpai adanya anemia microcytic hypochromic ringan (Hb 12.6 g/dL, MCH 22 pg, MCV 68 fL)²¹˒²³.
3. Hemoglobin H disease (--/-α)
Delesi dari 3 gen-α. Ciri hematologis ditandai adanya akumulasi dari rantai globin-β yang mudah larut membentuk tetramer β₄ yang disebut HbH yang pada pemeriksaan pewarnaan supravital dijumpai adanya badan inklusi (Heinzs bodies). Diagnosis penyakit HbH dengan menggunakan pemeriksaan hemoglobin Elektroforesis. Penyakit HbH memiliki gejala anemia hipokromik mikrositik dengan Hb 8-10 g/dL. Pada pemeriksaan fisik dijumpai adanya pembesaran Hepar dan spleen. Adanya anemia yang berat dapat disebabkan oleh
kekurangan asam folat, infeksi akut, paparan stres oksidatif, dan kehamilan. Pengobatan terdiri dari asam folat suplemen (5 mg/hari) dan transfusi darah⁸.
4. Hydrops Fetalis(--/--)
Merupakan delesi dari ke 4 rantai α. Janin yang terkena akan meninggal di dalam kandungan pada trimester kedua atau trimester ketiga kehamilan atau tidak lama setelah lahir³⁰. Keadaan ini terjadi pada thalassemia-α⁰ homozigot, tidak terbentuknya ke empat rantai globin-α. Pada keadaan ini hemoglobin fetus (HbF atau α22) tidak terbentuk pada masa janin dalam kandungan yang mengakibatkan rantai globin- yang tidak mendapatkan pasangan selanjutnya akan mengalami agregasi membentuk tetramer 4 yang disebut Hb-Bart’s. Terjadi anemia yang berat , mengalami oedem yang luas, ascites, efusi pleura, dan efusi pericardial²˒¹². Pada pemeriksaan apusan darah tepi banyak dijumpai immature red cell , hipokrom, mikrositer, gambaran sel darah merah anisopoikilositosis².
2.1.3.1. Thalassemia-β
Terdapat lebih dari 200 mutasi thalassemia-β yang telah diakui dan terjadi dalam berbagai kelompok etnis . Thalassemia-β umumnya terdapat di daerah Mediterania, di anak benua India di Asia Tenggara dan umumnya pada orang-orang keturunan Afrika³¹˒³²˒³³. Mutasi thalassemia-β dibagi menjadi dua Kategori: Thalassemia-β⁰ (beta zero) dan Thlassemia-β⁺ (beta plus)³¹.
Thalassemia-β dapat terjadi oleh karena hilangnya atau berkurangnya produksi dari rantai globin-β, dapat dibagi menjadi:
1. Thalassemia-β minor (trait)
Pada β-thalassemia trait kelainan terjadi oleh karena ketidakseimbangan sintesa rantai globin-β. Pada thalassemia-β minor (trait)\ tidak mengalami anemia yang berat, tapi pada pemeriksaan darah lengkap di jumpai mikrositer (MCV<80 fl) dan hipokrom (MCH<27 pg).
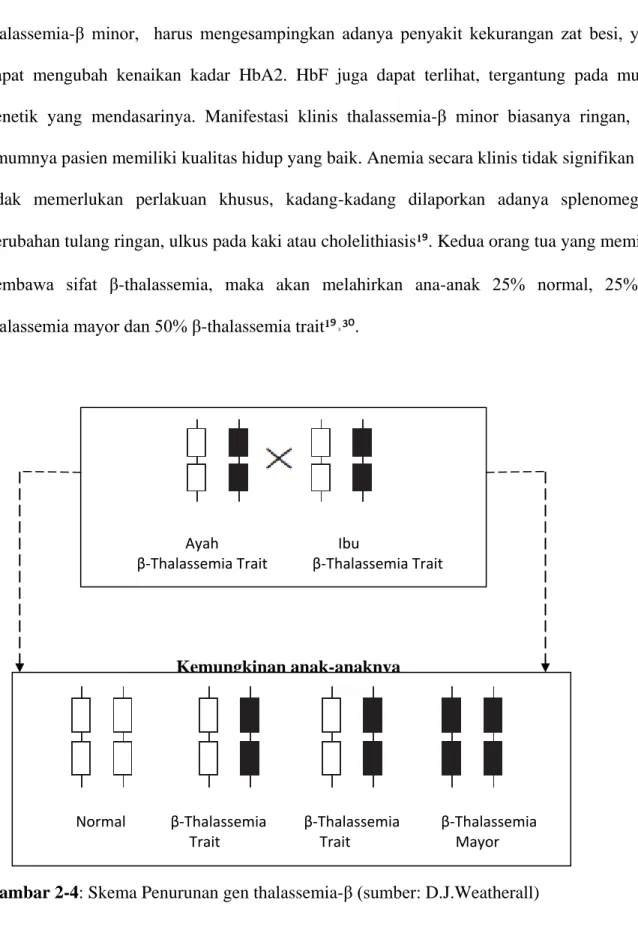
Pemeriksaan hemoglobin elektroforesis di jumpai peningkatan dari Hb A2 (>3,5%), namun pada thalassemia-α trait nilai HbA2 dapat normal atau menurun³⁰. Dalam membuat diagnosis thalassemia-β minor, harus mengesampingkan adanya penyakit kekurangan zat besi, yang dapat mengubah kenaikan kadar HbA2. HbF juga dapat terlihat, tergantung pada mutasi genetik yang mendasarinya. Manifestasi klinis thalassemia-β minor biasanya ringan, dan umumnya pasien memiliki kualitas hidup yang baik. Anemia secara klinis tidak signifikan dan tidak memerlukan perlakuan khusus, kadang-kadang dilaporkan adanya splenomegaly, perubahan tulang ringan, ulkus pada kaki atau cholelithiasis¹⁹. Kedua orang tua yang memiliki pembawa sifat thalassemia, maka akan melahirkan ana-anak 25% normal, 25% β-thalassemia mayor dan 50% β-β-thalassemia trait¹⁹˒³⁰.
Kemungkinan anak-anaknya
Gambar 2-4: Skema Penurunan gen thalassemia-β (sumber: D.J.Weatherall) Ayah Ibu
β-Thalassemia Trait β-Thalassemia Trait
Normal β-Thalassemia β-Thalassemia β-Thalassemia Trait Trait Mayor
2. Thalassemia-β Intermedia
Hampir 10% pasien thalassemia-β mengalami thalassemia-β intermedia (TI). Genetik dari kelompok ini mungkin memiliki homozigot talasemia-δβ atau homozygous atau heterozygous thalassemia β⁰ dan atau mutasi thalassemia-β⁺ . Pada TI mengalami anemia hemolitik yang sedang, dengan mempertahankan Hb >7 g/dl tanpa dukungan transfusi. Dalam penggunaan transfusi dibagi thalassemia-β intermedia dari thalassemia-β mayor. Ketika kebutuhan transfusi mencapai > 8 unit pertahun maka diklasifikasikan sebagai thalassemia-β mayor. Gejala klinis yang tampak pada TI biasanya terjadi pada umur 2-4 tahun. Gejalanya dapat berupa anemia, hiperbilirubinemia, dan hepatospleenomegali. Memiliki pertumbuhan yang lebih baik. Pada beberapa anak TI, walaupun Hb>7g/dl dapat mengalami kegagalan dalam pertumbuhan ,kurus yang tidak dapat kembali seperti semula kecuali apabila dilakukan transfusi reguler sebelum umur 6 atau 7 tahun ¹⁹˒²⁸.
3. Thalasemia Mayor
Thalassemia-β mayor selalu disebut anemia Cooley, anemia Mediterranean dan anemia Jaksch menunjukkan bentuk penyakit yang homozigot ataupun yang heterozigot ditandai dengan gejala anemia berat (1-7 g/dL), hemolisis dan inefektif eritropoesis yang berat. Manifestasi yang muncul pada masa anak-anak dapat terjadi anemia yang berat, ikterus, pertumbuhan terhambat, aktivitas menurun dan sering tidur. Hepatosplenomegaly dengan tanda awal dari wajah thalassemia biasanya ditemukan¹⁹. Pada pemeriksaan hapusan darah tepi dijumpai poikilositosis, mikrositosis, hipokrom, target sel, basophilic stipling, pappenheimer bodies (siderotic granules) dan retikulositosis dengan peningkatan Nucleated Red cells³².
Tabel 2-1 : Klasifikasi Thalassemia-β². 2.1.4. Epidemiologi
Thalassemia awalnya dianggap penyakit yang terdapat pada wilayah Mediterania, namun sekarang telah terjadi secara luas di seluruh penjuru dunia. Thalassemia telah dijumpai di Eropa Selatan dari Portugal ke Spanyol, Italia dan Yunani, serta di sejumlah negara Eropa Tengah dan bagian dari bekas Uni Soviet. Thalassemia juga dijumpai di Timur Tengah melalui Iran, Pakistan, India, Bangladesh, Thailand, Malaysia, Indonesia dan selatan Cina, serta negara-negara di pantai utara Afrika dan Amerika Selatan¹³ .
Migrasi penduduk dan adanya perkawinan campuran antara berbagai kelompok etnis telah mengembangkan thalassemia di hampir setiap negara di dunia, termasuk Eropa Utara di mana sebelumnya thalassemia ternyata tidak ada dan sekarang thalassemia menjadi masalah kesehatan umum utama. Diperkirakan 1.5% populasi dunia atau sekitar 80–90 juta orang carrier β-thalassemia, dengan sekitar 60.000 anak lahir pertahun memiliki kasus thalassemia, yang sebagian besar terjadi di dunia yang sedang berkembang¹³. Hemoglobin E-β-thalassemia salah satu hemoglobinopati paling sering dijumpai diseluruh dunia. Insiden HbE banyak terjadi pada 60 populasi di daerah Asia Tenggara. Di daerah pantai Amerika
Utara, prevalensi berkembang pesat. Penyakit α- talasemia sekarang juga sudah banyak dilaporkan. HbH, Hb Constants Spring , dan homozigot α-thalassemia mempengaruhi sekitar satu juta orang di seluruh dunia. 3% dari populasi di dunia (sekitar 150 juta orang ) memiliki gen carrier β-thalassemia³.
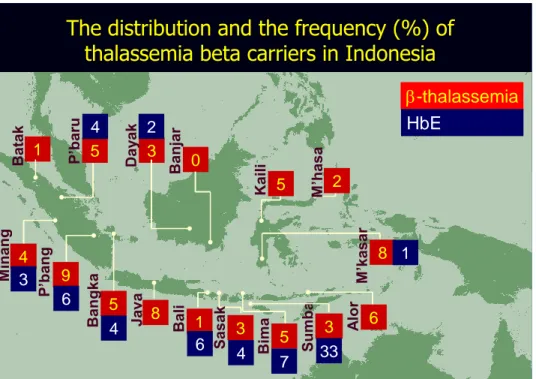
The distribution and the frequency (%) of
thalassemia beta carriers in Indonesia
1 Bat ak 9 P ’ban g 3 Day ak 0 Banjar 1 Bali 3 Su mb a 8 M ’k as ar 5 B an gka 8 Ja va 6 Alor 5 Bima 3 Sas ak 4 M inan g 5 P ’baru 5 Kaili M’hasa 2
A.S. Sofro & F. Lanni, University of Gajah Mada 4 6 4 3 6 4 33 7 1 2 -thalassemia HbE
Gambar 2-5: Distribusi dan frekuensi (%) carriers β-Thalassemia di Indonesia
(sumber: Thalassemia International Federation)
2.1.5. Diagnosis
Diagnosis thalassemia ditegakkan berdasarkan kriteria:
2.1.5.1. Anamnese
Dalam mendiagnosa thalassemia sangat penting mengetahui tentang riwayat penderita dan keluarga, karena ada beberapa populasi dengan ras etnik tertentu memiliki frekuensi yang tinggi untuk jenis gen abnormal thalassemia.
2.1.5.2. Pemeriksaan fisik
Pada pemeriksaan fisik penderita thalassemia dapat dijumpai adanya tanda pucat yang menunjukkan adanya anemia, ikterus adanya pembesaran organ seperti splenomegali, hepatomegali, dan skeletal formation³⁰˒³⁴.
2.1.5.3. Pemeriksaan Laboratorium
Pemeriksaan laboratorium meliputi, pemeriksaan darah lengkap (complete blood count/CBC), khususnya memeriksa nilai eritrosit rerata seperti Mean Corpuscular Volume (MCV), Mean corpuscular hemoglobin (MCH), Mean Corpuscular Hemoglobin Concentration (MCHC), Red Blood Cell Distribution Width (RDW). Pada pasien thalassemia-α maupun thalassemia-β menunjukan nilai MCV dan MCH yang rendah (Mikrositer hipokrom) dan mengalami anemia. Pada kasus β-thalassemia trait mengalami anemia yang ringan³⁵˒³⁶. Pemeriksaan laboratorium pada thalassemia diperlukan juga evaluasi sediaan hapusan darah tepi, badan inklusi HbH serta analisa hemoglobin dengan pemeriksaan hemoglobin elektroforesis dengan menilai kadar HbA2 dan kadar HbF. Kuantitasi HbA2 yang meningkat >3,5% mengidentifikasi suatu β-thalassemia trait ¹⁰. Analisa hemoglobin selain hemoglobin elektroforesis yaitu dengan menggunakan HPLC. Mutasi yang terjadi sehingga mengakibatkan diagnosis negatif palsu, maka pemeriksaan analisa genetik sangat diperlukan.
2.1.6 Patofisiologi
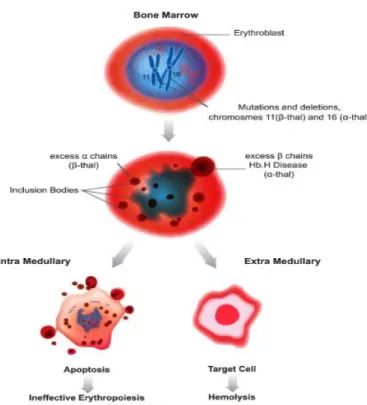
Patofisiologi yang mendasari antara jenis thalassemia hampir sama, ditandai dengan penurunan produksi hemoglobin dan sel darah merah (RBC) , adanya kelebihan rantai globin yang tidak efektif, akan menyebabkan bentuk homotetramers yang tidak stabil sehingga memicu terjadinya heinz body. Alfa homotetramers pada β-talasemia lebih tidak stabil daripada β-homotetramers di α-talasemia dan sebelumnya akan terbentuk presipitasi pada RBC, menyebabkan kerusakan sel darah merah dan hemolisis yang berat oleh karena eritropoesis yang tidak efektif serta hemolisis ekstramedular¹⁹. Pada β-thalassemia
patofisiologinya berdasarkan karena berkurang atau hilangnya rantai globin-β yang akan mengakibatkan berlebihnya rantai-α. Maka akan terjadi penurunan produksi hemoglobin dan ketidak seimbangan rantai globin. Ini akan mengarah pada penurunan dari volume hemoglobin (MCH) dan volume eritrosit (MCV)³. Pada thalassemia-β yang berat, eritropoesis yang tidak efektif terjadi di sum-sum tulang akan meluas ke tulang-tulang normal dan menyebabkan distorsi dari tengkorak kepala, tulang wajah dan tulang panjang. Aktivitas proliferasi eritroid di ekstramedular, akan menyebabkan limfadenopati, hepatosplenomegali, dan pada beberapa kasus terjadi tumor extramedular¹⁹˒³⁷.
Tidak efektifnya eritropoesis yang berat pada anemia kronis dan hipoksia dapat menyebabkan peningkatan absorbsi besi pada saluran pencernaan. Penderita thalassemia homozigot atau pun thalassemia-β heterozygot akan meninggal pada usia 5 tahun karena anemia yang berat. Namun transfusi menyebabkan penumpukan besi yang progressif oleh karena ekskresi yang tidak baik¹⁹.
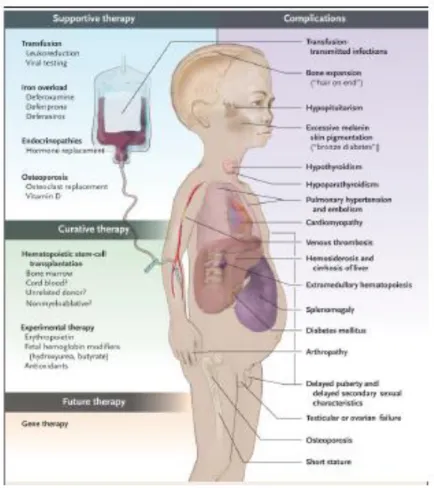
2.1.7 Komplikasi
Anemia pada pasien thalassemia umumnya berat disebabkan oleh karena tidak efektifnya eritropoesis dan mengakibatkan hematopoesis ekstramedular pada hati, limpa, dan tempat yang lain seperti paravertebral mass. Transmisi infeksi dapat terjadi oleh karena transfusi (contohnya hepatitis B dan C) . Besi yang berlebihan dari transfusi menyebabkan hemosiderosis dan meningkatnya penyerapan besi di saluran pencernaan. Besi yang mengendap di jantung, hati, dan kelenjar endokrine akan menyebabkan kerusakan yang berat. Aritmia dan gagal jantung, merupakan penyebab utama yang dapat menyebabkan pasien thalassemia meninggal. Terganggunya pertumbuhan dan perkembangan oleh karena besi akan menyebabkan kerusakan sumbu pituitary yang dapat terjadi tertundanya pertumbuhan pubertas dan perkembangan seksual. Hampir 90% dari pasien thalassemiaa mayor memiliki massa tulang yang rendah yang dikaitkan dengan tingginya kejadian fraktur¹⁹. Dapat terjadi peningkatan resiko thromboembolik oleh karena adanya berbagai kelainan trombosit dan faktor-faktor pembekuan². Telah banyak di laporkan komplikasi thromboembolik pada pasien thalassemia, menggambarkan adanya thrombotik di otak ³⁷˒³⁸.
Gambar 2-7: Komplikasi yang terjadi pada thalassemia³³.
2.2 Hemoglobin A2 (HbA2)
Manusia dewasa memiliki darah normal yang terdiri dari fraksi hemoglobin HbA, HbF, dan HbA2. HbA merupakan komponen mayor dari fraksi hemoglobin α2β2, dengan kadarnya 96,8%-97,8%, sedangkan komponen minor terdiri dari rantai globin α2ϒ2 (HbF) dengan kadar <0,5%, dan α2δ2 (HbA2) dengan kadarnya 2,2%-3,2%³⁰. Peningkatan Hemoglobin A2 merupakan parameter yang paling signifikan dalam mengidentifikasi β-thalassemia trait³⁹. Distribusi HbA2 pada 200 orang sehat dengan menggunakan alat elekroforesis dengan media celulosa acetate diperoleh HbA2 berkisar 1,5-3,5% (Weatherall et al,1971). Cutoff yang banyak digunakan peneliti sebagai batas atas HbA2 pada populasi sehat adalah 3,5%, dan digunakan juga selama penelitian ini untuk studi perbandingan. Selain pemeriksaan CBC dalam mendiagnosa thalassemia-β trait sangat penting dilakukan pemeriksaan kuantitasi dari HbA2, sehingga diperlukan presisi yang baik dalam metode kuantitasi HbA2. Ada beberapa
metode dalam pemeriksaan HbA2 yaitu Hemoglobin elektroforesis dengan media sellulose asetat, kromatografi kolom mikro, high performance liquid chromatography (HPLC) dan elektroforesis kapiler. International Committe for Standardization in haematology (ICSH) menganjurkan metode terpilih dengan kromatografi mikro kolom karena memiliki ketelitian yang baik dengan CV<4%. Elektroforesis kapiler (CE) mampu membedakan hemoglobin E (HbE) dari HbA2, sehingga dapat membedakan dengan baik kuantifikasi HbA2 pada pasien dengan HbE⁴⁰.
2.3 Kerangka Konsep
Kadar MCV<80fl Kadar MCH <27 pg
Kadar HbA2 >3,5% Β-thalassemia trait