92
E
VOLUTION OF THEFAD2-1
FATTY ACID DESATURASE5
9
UTR
INTRON AND THE MOLECULAR SYSTEMATICSOF
G
OSSYPIUM(M
ALVACEAE)
1Q
INGL
IU,
2,5C
URTL. B
RUBAKER,
3A
LLANG. G
REEN,
2D
ONR. M
ARSHALL,
4P
ETERJ. S
HARP,
4 ANDS
URINDERP. S
INGH22CSIRO Plant Industry, GPO Box 1600, Canberra, ACT 2601, Australia;
3Centre for Plant Biodiversity Research, CSIRO Plant Industry, GPO Box 1600, Canberra, ACT 2601, Australia; and 4University of Sydney, Plant Breeding Institute, Cobbitty, PMB11, Camden, NSW 2570, Australia
The FAD2-1 microsomalv-6 desaturase gene contains a large intron (;1133 bp [base pairs]) in the 59untranslated region that may participate in gene regulation and, in Gossypium, is evolving at an evolutionary rate useful for elucidating recently diverged lineages. FAD2-1 is single copy in diploid Gossypium species, and two orthologs are present in the allotetraploid species. Among the diploid species, the D-genome FAD2-1 introns have accumulated substitutions 1.4–1.8 times faster than the A-genome introns. In the tetra-ploids, the difference between the D-subgenome introns and their A-subgenome orthologs is even greater. The substitution rate of the intron in the D-genome diploid G. gossypioides more closely approximates that of the A genome than other D genome species, highlighting its unique evolutionary history. However, phylogenetic analyses support G. raimondii as the closest living relative of the D-subgenome donor. The Australian K-genome species diverged 8–16 million years ago into two clades. One clade comprises the sporadically distributed, erect to suberect coastal species; a second clade comprises the more widely spread, prostrate, inland species. A comparison of published gene trees to the FAD2-1 intron topology suggests that G. bickii arose from an early divergence, but that it carries a G. australe-like rDNA captured via a previously undetected hybridization event.
Key words: cotton; FAD2-1; fatty acid desaturase; Gossypium; intron; Malvaceae; polyploidy; reticulate evolution.
All higher plants contain one or more microsomalv-6 de-saturase(s) that insert a double bond between carbons 12 and 13 of monounsaturated oleic acid to generate polyunsaturated linoleic acid. This enzyme is mainly responsible for the pro-duction of the polyunsaturated fatty acids that are integral components of plant cellular membranes and of storage lipids in many vegetable oils (Shanklin and Cahoon, 1998). In
Gos-sypium, the microsomal v-6 desaturase family comprises at
least two distinct members in diploid species and perhaps as many as five in the allotetraploid species (Liu et al., 1996). Of particular interest is one member of this gene family,
FAD2-1, which is encoded by at least two copies in the tetraploid
cotton species G. barbadense and G. hirsutum (ghFAD2-1), and by a single copy in the diploid cotton species G.
arbo-reum, G. raimondii, and G. robinsonii (Liu et al., 1999). FAD2-1 is highly expressed and seed-specific and,
there-fore, is probably the main contributor of the polyunsaturated fatty acids in the seed oil of cultivated cottons (Liu et al., 1999). In addition to the three histidine boxes that are typical of all membrane-bound desaturases, the ghFAD2-1 gene con-tains a stretch of six contiguous glycine residues in the C-terminus of the open reading frame. Moreover, comparisons of genomic and cDNA clones encoding the ghFAD2-1 gene revealed a single large intron (;1133 bp [base pairs]) in the 59 untranslated region (UTR) located 9 bp upstream from the putative translation start site (Liu et al., 1997). Preliminary examination of the FAD2-1 gene from five species (G.
arbo-reum, G. barbadense, G. hirsutum, G. raimondii, and G. ro-binsonii) revealed that the size and position of the intron were
conserved. Sequence comparisons also suggested that the
1Manuscript received 3 September 1999; revision accepted 3 March 2000.
This work was supported by grants CSP78C and CSP85C from the Cotton Research & Development Corporation.
5Author for reprint requests ([email protected].)
FAD2-1 intron may be evolving at a quick enough rate for
inferring evolutionary relationships among recently diverged lineages and, in this regard, could be particularly useful for elucidating evolutionary pathways among the 17 Gossypium species indigenous to Australia, a group whose evolutionary history remains unresolved (Seelanan et al., 1999).
The current evolutionary understanding of the 17 Australian
Gossypium species is based on morphological and cytological
comparisons, and the phylogenetic analyses of three nucleotide sequences derived from the rpl16 intron (1155 bp), the 18S– 26S rDNA internal transcribed spacer (ITS: 688 bp), and a portion of an alcohol dehydrogenase gene (AdhD: 1600 bp) (Seelanan et al., 1999). These analyses confirmed hypotheses regarding the basal divergences on the Australian continent but provided little resolution of the evolutionary relationships among the 12 species indigenous to the Kimberley plateau of northwestern Australia. The nuclear gene topologies were also incongruent regarding the evolution of G. bickii, which has a biphyletic ancestry. Gossypium bickii captured a G.
sturtian-um-like chloroplast earlier in its evolutionary history, but to
date there is no evidence that this was accompanied by nuclear introgression (Wendel, Stewart, and Rettig, 1991). The topo-logical incongruencies may point to the first evidence that hy-bridization and introgression also altered the composition of
G. bickii’s nuclear genome, or that G. bickii experienced a
second and heretofore undescribed evolutionary reticulation. To provide an evolutionary context for investigations into the regulatory role of ghFAD2-1 intron and to refine our un-derstanding of the evolution of the Australian Gossypium spe-cies, the FAD2-1 intron was cloned and sequenced from 31
Gossypium species. Each major geographic region within the
New World allotetraploids. The resultant data allowed us to address the following questions: (1) Does the intron occur in all the Gossypium FAD2-1 genes? (2) Are the two copies in the allotetraploid species orthologs, inherited from their A and D genome progenitors, respectively? and (3) Does this intron contain sufficient phylogenetic signal to resolve the evolution-ary pathways among the Australian Gossypium species and the ambiguities regarding the evolution of G. bickii?
MATERIALS AND METHODS
The microsomalv-6 desaturase FAD2-1 intron was amplified and cloned from 39 accessions of 31 Gossypium species (Table 1). Multiple accessions of G. australe, G. bickii, G. nelsonii, G. robinsonii, and G. sturtianum were assayed to strengthen inferences regarding the basal divergences among the Australian Gossypium species. Pairs of putatively orthologous clones were sequenced from each of the five tetraploid species. The two A-genome species and five representative D-genome species were included to confirm the in-ferred subgenomic origin of the clones of the tetraploid species, including the two D-genome species nominated as most likely to be sister to the D-sub-genome (Endrizzi, Turcotte, and Kohel, 1985; Wendel and Albert, 1992; Wen-del, Schnabel, and Seelanan, 1995).
Total genomic DNA was extracted following Paterson, Brubaker, and Wen-del (1993) and further purified by CsCl gradients following Sambrook, Fritsch, and Maniatis (1989). The entire 59UTR intron was amplified using primers that flanked the predicted splice site. The upstream primer (S1: 59-CCTGGCGTTAAACTGCTTTC-39) is located at 44–63 bp downstream of the transcription start site in the 59UTR and the downstream primer (A1: 59-GCATAGGTCATGGACCACGT-39) is located at 239–258 in the coding re-gion (exon2) of ghFAD2-1 (EMBL accession X97016). The 50-mL polymer-ase chain reactions (PCRs) contained 200mmol/L dNTPs, 1X PE Applied Biosystems (Scoresby, VIC, Australia) PCR buffer, 20 pmol of each primer, 10 ng genomic DNA, and 1 unit of Taq DNA polymerase. PCRs started with a 2-min denaturation at 948C, followed by 30 cycles of 948C for 1 min, 568C for 1 min, and 728C for 1 min, and finished with 10-min final extension at 728C. PCR products were purified with WizardtPCR Preps DNA Purification System (Promega; Annandale, NSW, Australia) and cloned into Tt-vector (Promega) according to manufacturer’s instructions. Plasmids were isolated following Sambrook, Fritsch, and Maniatis (1989), and the DNA sequences were determined using the PRISMy kit (PE Applied Biosystems) on an ABI373 DNA Sequencer.
Sequence analysis—Sequences were initially aligned using GCG-pileup
(Wisconsin Package Version 9.1, Genetics Computer Group [GCG], Madison, Wisconsin, USA) and then adjusted manually. Individual sequences have been submitted to EMBL (Table 1); the sequence alignment was also submitted to EMBL (Accession DS41945). Mega 1.01 (Kumar, Tamura, and Nei, 1993) was used to characterize the sequences and compute pairwise Jukes-Cantor distances (Jukes and Cantor, 1969).
Topologies were inferred heuristically using the GCG implementation of PAUP (Wisconsin Package Version 9.1, Genetics Computer Group [GCG], Madison, Wisconsin, USA) using parsimony or distance (minimum evolution) as the optimality criterion. In both cases, starting trees were acquired by step-wise addition (simple), ten trees were held at each step, and the TBR algo-rithm was used for branch swapping using steepest descent. Gaps were treated as missing data, and potentially informative indels were recoded as binary characters and included in some analyses. Distance-optimized topologies were initially inferred using the Jukes-Cantor (Jukes and Cantor, 1969) model of nucleotide substitution, however, because transition/transversion ratios were generally,2 but the frequencies of the four nucleotides deviated substantially from equality, the Tajima-Nei (Tajima and Nei, 1984) estimator was also used (Kumar, Tamura, and Nei, 1993). Sites containing gaps and regions of am-biguous homology were ignored. Negative branch lengths were set to zero. The model for substitution rate variation across sites was determined by the gamma distribution (shape parameter set equal to 0.5). As a measure of clade ‘‘strength,’’ Autodecay, in association with PAUP 3.1 for Macintosh (Eriksson
and Wikstro¨m, 1995; Swofford, 1991), was used to determine the length of the shortest tree in which each clade failed to appear (Bremer, 1988; Dono-ghue et al., 1992). Relative rate tests followed Tajima (1993) using Tajima93 (see Seelanan et al., 1999).
RESULTS
Comparison of the cDNA sequence of the G. hirsutum
ghFAD2-1 gene and its corresponding genomic clone
con-firmed the presence of a single large intron in the 59 untrans-lated region (UTR) (Liu et al., 1997, 1999). The intron is ;1133 bp and is located 9 bp upstream of the translation ini-tiation site in G. hirsutum. This is strikingly similar to the microsomalv-6 desaturase in Arabidopsis thaliana, which has a 1130-bp intron located 4 bp upstream of the translation ini-tiation site (Okuley et al., 1994). In contrast, the Glycine max
FAD2-1 has a much smaller intron of 320 bp located 4 bp
downstream of the translation initiation site (Liu et al., 1997). Attempts to align these three intron sequences revealed nu-merous sequence dissimilarities and no obvious regions of conservation.
Using the primers developed for G. hirsutum, the intron was amplified from the 39 Gossypium accessions (Table 1). The diploid Gossypium species contained a single copy of the
FAD2-1 gene, and in each case the gene contained the 59UTR
intron. The five allotetraploid species contained two distinct
FAD2-1 genes and each contained an intron (subgenus Kar-pas; Table 1). All of the introns started with GT and ended
with AG, consistent with the plant consensus 59and 39exon/ intron boundaries (Simpson and Filipowicz, 1996). The introns had a mean GC content of 24%, and the 154 bp of exon 2 had a mean GC content of 55%. The length of the introns ranged from 1065 (G. australe-2) to 1166 bp (G.
gossypioi-des). The G-genome species, G. raimondii, and the tetraploid
A- and D-subgenome orthologs had mean intron lengths below 1120 bp, while the A-, C-, D-, E-, and K- genome species had mean intron lengths greater than 1130 bp. The introns con-tained 14 simple sequence repeat regions with more than five repeat units in at least one accession: 12 (T)n, 1 (CT)n, and 1
(AAG)n(individual accession data available from
correspond-ing author).
Phylogenetic analysis—The final aligned length of the
an-alyzed matrix was 1406 bp. The anan-alyzed sequences start with the first nucleotide of the intron and end with the 154th nu-cleotide of the second exon. Twenty-eight of the insertion/ deletion events (1–57 bp) inferred from this alignment were potentially phylogenetically informative and were coded as bi-nary characters. The 154 nucleotides of exon 2 aligned without gaps. Simple sequence repeat regions were excluded. Homol-ogy assessments in several other short regions were ambiguous and also excluded. Of the final aligned length of 1406 bp, 221 bp were excluded from phylogenetic analyses. Considering only the 1185 nucleotide positions used in the phylogenetic analyses, 344 were variable, of which 169 were parsimony informative: 319 variable sites occurred in the intron, of which 158 were parsimony informative; 25 variable sites occurred in 154 bp of the 59end of the exon, of which 11 were parsimony informative.
char-TABLE1. Gossypium accessions assayed. Full provenance details available from C. L. Brubaker. Genome designations follow Stewart (1995); taxonomy follows Fryxell (1992).
Taxa Genome Accession no. Locality/Cultivar EMBL no.a
Subgenus Sturtia (R. Brown) Todaro Section Sturtia
G. robinsonii F. Mueller 1 C CAT #1364 Nindethana Seed Service, WA, Australia
EMBL-AJ244884 2 C GOS-5170 WA: Australia (MillStream
Na-tional Park)
EMBL-AJ244885 G. sturtianum J. H. Willis 1 C s.n. NT: Australia (Hermansburg;
Col-lected by P. Abell)
EMBL-AJ244886 2 C GOS-5297 NT: Australia (Glen Helen
Sta-tion)
EMBL-AJ244887
3 C GOS-5076 NSW: Australia (Narrabri) EMBL-AJ244888
Section Grandicalyx (Fryxell) Fryxell
G. costulatum Todaro K GOS-5378 Yampi Peninsula (WA: Australia) EMBL-AJ244889 G. cunninghamii Todaro K GOS-5309 Cobourg Peninsula (WA: Australia) EMBL-AJ244890 G. enthyle Fryxell, Craven, &
Stewart
K GOS-5176 Mitchell River Basin (WA: Aus-tralia)
EMBL-AJ244891 G. exiguum Fryxell, Craven, &
Stewart
K GOS-5347 King Edward River (WA: Australia) EMBL-AJ244892 G. londonderriense Fryxell,
Cra-ven, & Stewart
GOS-5191 Drysdale River Mouth NE of Kal-umburu (WA: Australia)
EMBL-AJ244893 G. marchantii Fryxell, Craven, &
Stewart
K GOS-5193 Bougainville Peninsula (WA: Aus-tralia)
EMBL-AJ244894 G. nobile Fryxell, Craven, &
Stewart
K GOS-5196 Carson River Station (WA: Aus-tralia)
EMBL-AJ244895 G. pilosum Fryxell K GOS-5203 Northern end of Mitchell Plateau
(WA: Australia)
EMBL-AJ244896 G. populifolium (Bentham) G.
Mueller ex Todaro
K GOS-5210 Augustus Island (WA: Australia) EMBL-AJ244897 G. pulchellum (C. A. Gardner)
Fryxell
K GOS-5204 Vansittart Bay (WA: Australia) EMBL-AJ244898
G. rotundifolium Fryxell, Craven,
& Stewart K GOS-5030 16 km North of Broome towards
Beagle Bay (WA: Australia)
EMBL-AJ244899 G. species novum (see Stewart,
Craven, and Wendel, 1997)
K GOS-5223 Cape Talbot (WA: Australia) EMBL-AJ244900
Section Hibiscoidea Todaro
G. australe F. Mueller 1 G CAT #1363 Nindethana Seed Service, WA EMBL-AJ244901
2 G GOS-5041 Carawine Gorge (WA: Australia) EMBL-AJ244902
3 G PI-499756 Ormiston Gorge (NT: Australia) EMBL-AJ244903 G. bickii Prokhanov 1 G GOS-5338 Urandangie Road S of Barkly
Hwy (QLD: Australia)
EMBL-AJ244904 2 G PI-464843 Supplejack Station (NT: Australia) EMBL-AJ244905
3 G GOS-5048 Alice Springs (NT: Australia) EMBL-AJ244906
G. nelsonii Fryxell 1 G GOS-5024 Richmond (QLD: Australia) EMBL-AJ244907 2 G PI-499783 Ormiston Gorge (NT: Australia) EMBL-AJ244908
Subgenus Houzingenia Fryxell Section Houzingenia
Subsection Houzingenia
G. trilobum (Mocin˜o & Sesse´ ex DC) Skovsted
D s.n. Mexico EMBL-AJ244909
Subsection Integrifolia (Todaro) Todaro
G. klotzschianum Andersson D GOS-5400 Gala´pagos Islands EMBL-AJ244910
Subsection Caducibracteolata Mauer
G. turneri Fryxell D GOS-5403 Mexico EMBL-AJ244911
Section Erioxylum (Rose & Stan-dley) Prokh.
Subsection Selera (Ulbrich) Fryxell
G. gossypioides (Ulbrich) Standley D CPI-138644 Mexico EMBL-AJ244912
Subsection Austroamericana Fryxell
G. raimondii Ulbrich D GOS-5330 Peru EMBL-AJ244913
Subgenus Gossypium Section Gossypium
Subsection Gossypium
G. arboreum L. A GOS-5264 India Type 9 EMBL-AJ244914
TABLE1. Continued.
Taxa Genome Accession no. Locality/Cultivar EMBL no.a
Subgenus Gossypium Section Gossypium
Subsection Pseudopambak (Prokh.) Fryxell
G. somalense (Gu¨ rke) J.B. Hutch-inson
E CPI-138057 Somalia, Kenya, Sudan EMBL-AJ244916
G. stocksii Masters E CPI-138058 Pakistan, Arabia, Somalia EMBL-AJ244917
Subgenus Karpas Rafinesque
G. barbadense L. AD s.n. Pima S7 EMBL-AJ244918 (A)
EMBL-AJ244919 (D)
G. darwinii Watt AD GOS-5399 Gala´pagos Islands EMBL-AJ244920 (A)
EMBL-AJ244921 (D)
G. hirsutum L. AD s.n. Deltapine-16 EMBL-AJ244922 (A)
EMBL-AJ244923 (D)
G. mustelinum Miers ex Watt AD GOS-5402 Brazil EMBL-AJ244924 (A)
EMBL-AJ244925 (D)
G. tomentosum Todaro AD GOS-5404 Hawaii EMBL-AJ244926 (A)
EMBL-AJ244927 (D)
aThe prefix EMBL- has been added to all the EMBL accession numbers to link the online version of American Journal of Botany to EMBL but
is not part of the actual accession number.
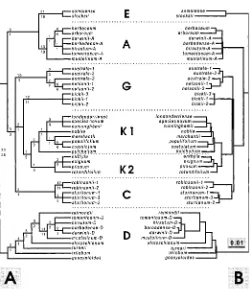
Fig. 1. Parsimony (A) and distance (B) optimized topologies of the FAD2-1 intron among 39 accessions of 3FAD2-1 Gossypium species. (A) Strict consensus tree of 103 equally parsimonious trees (517 steps; consistency index50.717, excluding uninformative characters; retention index50.912). Twenty-eight indels included in analysis as binary characters, otherwise gaps were treated as missing data. The number of unambiguous substitutions are indicated above branches; the decay index for each clade is indicated below each branch. (B) Single most parsimonious distance-optimized (Tajima and Nei, 1984) tree (485 steps ; consistency index50.688, excluding uninformative characters; retention index50.899)
acters, and retention indices (RI) of 0.901 (not illustrated). With the 28 indels included, an heuristic search returned 103 equally parsimonious trees (CI 5 0.717, excluding uninfor-mative characters; RI 5 0.912) (Fig. 1A). Strict consensus trees from analyses with and without the indels differed only in the resolution of the G. hirsutum-A/G. tomentosum-A/G.
barbadense-A/G. darwinii-A clade relative to the G. arbo-reum/G. herbaceum clade. When the indels were excluded,
these four A-subgenome introns collapsed to form a polytomy with the G. arboreum–G. herbaceum clade. With the indels included, the A-subgenome intron of these four species ap-peared as a sister clade to the G. arboreum/G. herbaceum clade (Fig. 1A).
Distance-optimized topologies were inferred from the pared sequence matrix using Jukes-Cantor (Jukes and Cantor, 1969) or Tajima-Nei (Tajima and Nei, 1984) models of nucleotide substitution. Both analyses returned a single and identical tree (CI50.688, excluding uninformative characters; RI50.899) (Fig. 1B). The distance- and parsimony-optimized topologies are congruent except for the placement of the E-genome clade and G. marchantii (Fig. 1).
In both distance- and parsimony-optimized topologies, all the diploid genome groups are resolved as monophyletic lin-eages except the Australian G genome. Bremer support for the monophyly of the A-, D-, E-, and K-genome clades is strong, whereas the monophyly of the C genome clade is weakly sup-ported (Fig. 1A). Because no outgroup was included, both to-pologies are midpoint rooted and no inferences regarding the basal divergence within Gossypium obtain from the figured topologies.
TABLE2. Relative rate test (2D; Tajima, 1993) of nucleotide substitution rates among the A-, D-, and E-genome species relative to G. costulatum, calculated using Tajima93 (see Seelanan, Schnabel, and Wendel, 1997). (P50.05.*.0.01.**.0.005.***). Exon sequence, regions of dubious homology, and indels were not included in analyses.
Taxon
Genome
designation 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
1 G. arboreum A
2 G. herbaceum A NS
3 G. barbadense A-AD * NS
4 G. darwinii A-AD NS NS NS
5 G. hirsutum A-AD NS NS NS NS
6 G. mustelinum A-AD * NS NS NS NS
7 G. tomentosum A-AD ** NS NS NS NS N/A
8 G. gossypioides D NS NS NS NS NS * *
9 G. klotzschianum D NS * ** * * *** *** NS
10 G. raimondii D NS * * * * ** *** NS NS
11 G. trilobum D NS * *** * * *** *** NS NS NS
12 G. turneri D NS * * * * ** *** NS NS NS NS
13 G. barbadense D-AD NS * *** * * *** *** NS NS NS NS NS
14 G. darwinii D-AD NS ** *** ** ** *** *** NS NS NS NS NS NS
15 G. hirsutum D-AD NS * *** * * *** *** NS NS NS NS NS NS NS
16 G. mustelinum D-AD NS * *** * * *** *** NS NS NS NS NS NS NS NS
17 G. tomentosum D-AD NS ** *** ** ** *** *** NS NS NS NS NS NS NS N/A NS
18 G. somalense E NS NS NS NS NS NS * NS NS NS NS NS NS NS NS NS NS
19 G. stocksii E NS * *** * * ** *** NS NS NS NS NS NS NS NS NS NS NS
The Australian Gossypium species (C, G, and K genomes) are also resolved as a monophyletic lineage in the parsimony-and distance-optimized topologies (Fig. 1). Within the Austra-lian clade there is a primary divergence into four subclades: (1) G. australe/G. nelsonii, (2) G. bickii, (3) the K genome, and (4) the C genome.
Among the K-genome species, two well-supported clades are evident (decay index of 3; Fig. 1). The first comprises G. species novum, G. costulatum, G. cunninghamii, G.
london-derriense, G. marchantii, G. nobile, G. populifolium, and G. pulchellum; the second comprises G. enthyle, G. exiguum, G. pilosum, and G. rotundifolium. Within the former, or K1 clade,
two subclades are evident, G. species novum/G. cunninghamii/
G. londonderriense, and G. costulatum/G. populifolium. The
relationship of G. nobile to these subclades is unresolved, and
G. marchantii is weakly supported as sister to the G. costu-latum–G. populifolium clade. Gossypium pulchellum is weakly
supported as the single extant ancestor of one lineage arising from the basal divergence in the K1 clade. The latter, or K2 clade, sees G. rotundifolium placed sister to an unresolved trichotomy comprising G. enthyle, G. exiguum, and G.
pilos-um.
The topological placement of G. bickii was unexpected. The two other G-genome species, G. australe and G. nelsonii, are strongly supported as sister species (decay index of 9), but G.
bickii resolves as sister to the K-genome clade (Fig. 1).
Be-cause this implied that the G genome was paraphyletic, further heuristic searches were undertaken to test the stability of this result. In the first instance, the data matrix was reanalyzed without the E-genome species (because of their own topolog-ical instability, discussed above), the C-genome species, or G.
australe and G. nelsonii. Subsequently, the E-genome species
in combination with the C-genome species or G. australe and
G. nelsonii were excluded. In all cases, distance- and
parsi-mony-optimized searches returned consensus topologies con-gruent with those illustrated in Fig. 1. The same topology was also recovered when the Australian species were analyzed with only the A-genome, D-genome, or the E-genome species.
The topological placement of the tetraploid sequences is
consistent with the original assessment that each tetraploid species contained a pair of orthologous loci. The putative D-subgenome sequences from the tetraploid species resolved as a monophyletic clade sister to G. raimondii within a clade of other diploid D-genome species. The putative A-subgenome sequences appear in a strongly supported A-genome clade (de-cay index of 13), but do not resolve as a monophyletic lineage. Gossypium mustelinum appears as basal to two sub-clades: (1) G. arboreum/G. herbaceum and (2) G. barbadense/
G. hirsutum/G. darwinii/G. tomentosum.
Relative rates of nucleotide substitution—Because the
bas-al divergence in Gossypium occurred between the ancestor of the A-, D-, E-, and AD-taxa and the ancestor of the C-, G-, and K-genome species (Wendel and Albert, 1992; Seelanan et al., 1997, 1999), the relative substitution rates (2D test; Tajima, 1993) among the Australian species were evaluated using G.
somalense, G. raimondii, or G. herbaceum as reference taxa.
The pared sequences were used, i.e., areas of ambiguous ho-mology were excluded, and regions with gaps in one or more taxa were excluded from all comparisons. All three analyses demonstrated that nucleotide substitution rates are largely ho-mogeneous among the species. Only G. species novum, G.
australe-3, G. londonderriense, and G. rotundifolium returned
significant chi-square tests for some species combinations (data not shown). These species were excluded from diver-gence time estimates (described below).
Conversely, homogeneity of substitution rates among the A-, D-, E-, and AD-genome species were tested using G.
costu-latum, G. nelsonii, G. robinsonii, or G. sturtianum,
respec-tively, as reference taxa. As above, the pared sequences were used to eliminate ambiguous homology assessments and sites with gaps were excluded from all comparisons. All four tests consistently demonstrated that nucleotide substitution rates are not homogeneous among these clades (Table 2). Particularly notable was that the A- and D-genome lineages, except G.
arboreum and G. gossypioides, have accumulated substitutions
at significantly different rates.
TABLE3. Mean Tajima-Nei distances (Tajima and Nei, 1984) between A or D species and the four Australian lineages. Exon sequence, regions of dubious homology, and indels were not included in the distance estimation.
Reference lineage
Mean Tajima-Nei distances (mean transition/transversion ratio)
A A subgenome G. herbaceum G. arboreum D D subgenome D genome diploids G. gossypioides
G. australe/G. nelsonii
TABLE4. Estimated divergence times among the Australian clades. A substitution rate of 0.721.731029nucleotide substitutions per site
per year was calibrated using the estimated age of divergence (10.5–21 mybp) between the C- and K-genome clades for AdhD reported by Seelanan et al. (1999). Estimated divergence times es-timated by dividing the mean Jukes-Cantor distance by twice the nucleotide substitution rate.
bSeelanan et al. (1999).
cBasal divergence among the K-genome species (see Fig. 1): K
ge-nome-15G. costulatum, G. cunninghamii, G. marchantii, G. nobile, and G. populifolium; K genome-2 5G. enthyle, G. exiguum, and G. pilosum.
dGossypium species novum, G. australe-3, G. londonderriense, and
G. rotundifolium were excluded (see text).
eSeelanan et al. (1999) calculated this figure assuming G. australe,
G. bickii, and G. nelsonii formed a monophyletic clade.
Tajima-Nei distances between the A- and between the D-ge-nome taxa and the four major Australian clades were calcu-lated (Table 3). Gossypium arboreum and G. gossypioides were considered separately, and G. australe-3, G. species no-vum, G. londonderriense, and G. rotundifolium were not in-cluded in the calculations. These comparisons demonstrate that D-genome introns accumulated ;1.8 more substitutions per site than did the A-genome introns. If these comparisons are partitioned into tetraploid subgenomic and diploid compo-nents, the A-subgenome taxa had a lower mean distance from the taxa in the four major Australian clades than did G.
ar-boreum and G. herbaceum, in contrast to the D-subgenome
taxa, which have a higher mean distance from the taxa in the four major Australian clades than is observed in the D diploid taxa. This suggests that the differences in nucleotide substi-tution rates between the A- and D-genome lineages have been magnified in the two polyploid lineages. These comparisons are consistent regardless of which of the Australian lineages are used as a point of reference.
The comparisons in Table 3 also illustrate why G. arboreum and G. gossypioides appeared as anomalies in the global rel-ative rate tests (Table 2). Both the A-genome diploids have higher mean Tajima-Nei distances relative to the Australian species than the A-subgenome homologs, however this differ-ence is most pronounced for G. arboreum, and it most closely approaches the mean Tajima-Nei distances between the D-ge-nome species and the Australian species. In contrast, the mean Tajima-Nei distances between the Australian species and D-genome diploid species are lower than those for the D-sub-genome homologs, and among the D-D-sub-genome diploids, G.
gos-sypioides has the lowest distance to the Australian species and
thus more closely approaches the nucleotide substitution rate of the A-genome species.
Divergence time estimates—Because substitution rates
among the Australian species were largely homogeneous, the mean Tajima-Nei distances were used to estimate the time since divergence among the major Australian clades. In each clade, the mean Tajima-Nei distances were calculated from all pairwise comparisons except for the taxa that returned signif-icant relative rate statistics (viz., G. species novum, G.
aus-trale-3, G. londonderriense, and G. rotundifolium). The
sub-stitution rate was calibrated using Seelanan et al.’s (1999) es-timate that the C genome diverged from the K genome 10.5– 21 million years before present (mybp). This returned an es-timated substitution rate of 0.7–1.7 3 1029substitutions per
site per year. Although the accuracy of this estimated substi-tution rate cannot be independently verified, it permits relative comparisons of divergence times among the major Australian clades derived from the chloroplast gene rpl16 and the nuclear gene AdhD (Seelanan et al., 1999).
The estimated divergence times derived from the FAD2-1 intron mostly are congruent with estimates derived from rpl16 and AdhD (Table 4). Comparisons are complicated because Seelanan et al. (1999) considered the G-genome species mono-phyletic, whereas G. bickii resolved as a clade in its own right here. Nonetheless, the estimated divergence times between the C-genome species and G. bickii (8.9–17.8 million years before present or mybp) or the G. australe/G. nelsonii clade (9.5– 18.9 mybp) overlaps previous estimated divergence between the C- and G-genome species of 8–15 mybp (Table 4). Simi-larly, the estimated time of divergence between the G. bickii and G. australe/G. nelsonii clades (8.4–16.9 mybp) overlaps the estimate of 8–15 mybp for the earliest divergence among the G-genome species based on analysis of AdhD sequences (Table 4). There are, however, two striking incongruities.
FAD2-1-based estimates for the basal divergence within the K
C genome are five- to tenfold and threefold older, respectively, than previously estimated (Table 4).
DISCUSSION
The A- and D-genome FAD2-1 introns are orthologs but are evolving at significantly different rate—In an earlier
pa-per, the size similarity between the two FAD2-1-specific re-striction fragments in G. barbadense and in G. hirsutum and the A-genome G. arboreum and the D-genome G. raimondii fragments suggested that the two allotetraploid FAD2-1 genes were A- and D-subgenome orthologs (Liu et al., 1999). The topological placements of the ten intron sequences from the five allotetraploid species support this interpretation (Fig. 1). Five of the allotetraploid sequences, one from each species resolved within a monophyletic D-genome lineage, sister to G.
raimondii; the other five resolved as part of a monophyletic
A-genome lineage. Support for these topological placements is strong. The A-subgenome sequences share 13 unambiguous substitutions with the two A-genome diploid species, and the D-subgenome sequences share 33 unambiguous substitutions with the D-genome diploid species.
Relative rate tests demonstrate that, except for G. arboreum and G. gossypioides (discussed below), the D-genome
FAD2-1 introns have accumulated FAD2-1.8 times the number of base
sub-stitutions than have the A-genome introns relative to the phy-logenetically equidistant Australian Gossypium species (Tables 2, 3). This difference is reflected in the relative branch lengths of the A- and D-genome clades in Fig. 1B. These differences are even greater among the A- and D-subgenome species (Ta-ble 3). For example, the mean Tajima-Nei distance of the D-genome diploids from G. bickii is 1.4 and 1.7 times greater than it is for G. herbaceum and G. arboreum, respectively, but the mean Tajima-Nei distance of the D-subgenome intron se-quences from G. bickii is 1.9 times greater than for the A-subgenome intron sequences. This pattern is consistent regard-less of the reference taxon used (Table 3). Small et al. (1998) made a similar observation based on relative rate tests of AdhC among all five Gossypium allotetraploid species, G. arboreum, and G. raimondii relative to G. robinsonii. Although nucleo-tide substitution rates for the A- and D-subgenome AdhC in-trons are roughly equal, the substitution rate in the D-subgen-ome exons is nearly five times greater than it is in the A subgenome. Overall, the D-subgenome AdhC sequences are accumulating substitutions about 1.8 times faster than their orthologs in the A subgenome (Small et al., 1998). The con-sequences of this differential rate of sequence evolution in phylogenetic reconstruction are evident in Fig. 1. The A su-bgenome lacks even one defining synapomorphy and is com-pletely unresolved except for one substitution that differenti-ates G. mustelinum from the other taxa. In contrast, the D subgenome is defined by 11 substitutions and is more fully resolved.
Small et al. (1998) suggest differential selection between the two subgenomes may partially account for different nucleotide substitution rates between orthologous genes in the same nu-cleus. While this certainly may play a role, the FAD2-1 intron analysis, which includes both A-genome species and six of the D-genome species suggests that the underlying mechanistic differences were active in the A- and D-subgenome progeni-tors and that these lineage differences have been exaggerated in the allopolyploids. In this regard it is notable that the G.
arboreum and G. raimondii AdhC sequences are also evolving
at significantly different rates, relative to each other, and rel-ative to their D-subgenome and A-subgenome orthologs, re-spectively (Small et al., 1998). More recent data suggest that the assumption that the A and D genomes are phyletically equidistant from the Australian species may be incorrect (J. F. Wendel, personal communication). However, even if the A-genome and the Australian species share a more recent com-mon ancestor than either does with the D genome, the phyletic distance between the A-genome/Australian and the D-genome lineages would have to be much greater than these data sug-gest it will be to account for the differences in the absolute number of nucleotide substitutions between the A- and D-ge-nome lineages (Wendel and Albert, 1992; Seelanan, Schnabel, and Wendel, 1997).
The exceptions to this differential rate of nucleotide substi-tution between the A and D subgenomes are G. arboreum and
G. gossypioides. The substitution rates of these two species,
run counter to their genomic compatriots. Gossypium
arbo-reum has the highest substitution rate among the A-genome
diploid and allopolyploid species, and G. gossypioides has the lowest rate among the D-genome species. Gossypium
gossy-pioides is accumulating substitutions only 1.2 times faster than G. arboreum compared to a general difference of 1.8 observed
between the A and D genomes (Table 3). This observation is intriguing because both of these species have unique evolu-tionary histories.
Gossypium arboreum is one of the four domesticated Gos-sypium species and is the only one for which a wild progenitor
has never been identified, existing only as a domesticated cul-tigen or feral derivatives of domesticated forms (Brubaker, Bourland, and Wendel, 1999). Although it is tempting to at-tribute some proportion of G. arboreum’s anomolous sequence substitution rate to the effects of human domestication, the degree to which human selection has altered rates of gene evolution in G. arboreum cannot be ascertained without the wild progenitor as a point of reference. Nonetheless, it is plau-sible that human selection has altered rates of sequence evo-lution in G. arboreum, an hypothesis worth testing in crop species for which clearly identified wild progenitors are avail-able.
Gossypium gossypioides is distinct among other D-genome
diploid species, because it contains a mosaic genome resulting from an ancient hybridization (Wendel, Schnabel, and Seelan-an, 1995). Despite the fact that there are no A-genome species extant in the New World, the G. gossypioides genome contains A-genome-specific dispersed repeats and a mosaic nuclear ri-bosomal DNA repeat that combines features of the A-genome rDNA repeat with those of the D-genome rDNA (Wendel, Schnabel, and Seelanan, 1995; Zhao et al., 1998). Whether there is a causal link between the G. gossypioides mosaic ge-nome and the tardy rate of sequence evolution in its FAD2-1 intron remains to be determined.
D-genome evolution and the origin of the New World allotetraploids—The topological placement of the
D-subgen-ome intron sequences sister to G. raimondii, while G.
gossy-pioides resolves as basal to the entire D-genome lineage, is
gossy-pioides experienced introgressive hybridization with the
pro-genitor of the allotetraploid A subgenome, either directly or indirectly via the nascent allotetraploid (Wendel, Schnabel, and Seelanan, 1995). Traditionally, G. raimondii is considered to be the extant D-genome species sister to the D-subgenome progenitor. This is based on leaf developmental patterns, seed hair type, vigor of F1hybrids with A-genome species,
chro-mosome homologies inferred from multivalent frequencies in synthetic hexaploid hybrids, and genetic segregation in syn-thetic allohexaploids (reviewed by Endrizzi, Turcotte, and Ko-hel, 1985). As comprehensive as this sounds, G. raimondii is a geographical outlier relative to the other D-genome diploid species. It is indigenous to Peru while the other species are found in Mexico (except for G. klotzschianum in the Gala´pa-gos). Thus some question the assumption that G. raimondii is the sister taxon to the D subgenome. This doubt is largely based on the suspicion that the allotetraploid lineage arose in Mexico (specifically the Isthmus of Tehuantepec) rather than South America (Wendel, Schnabel, and Seelanan, 1995). If this is true, G. raimondii is indeed an unlikely candidate despite its genomic and genetic similarities to the D subgenome, and, conversely, its putative sister taxon, G. gossypioides becomes a strong candidate, lying as it does closer to the Isthmus of Tehuantepec than any other D-genome species and carrying genomic evidence of introgression with the A-subgenome pro-genitor (Wendel, Schnabel, and Seelanan, 1995).
This reasoning benefits from parsimony as it assumes a sin-gle A3 D interspecific hybridization that resulted in the mo-saic G. gossypioides rDNA internal transcribed spacer and the evolution of an allotetraploid lineage rather than two hybrid-ization events between two geographically separated D-ge-nome species and a single A-geD-ge-nome taxon. Wendel, Schnabel, and Seelanan (1995) are also correct in questioning the reli-ability of multivalent frequencies and segregation ratios in synthetic hexaploids, the strongest but uncorroborated evi-dence rejecting G. gossypioides as the sister taxon of the D-subgenome progenitor. However, hypotheses that the allotetra-ploid lineage arose in Mexico and that G. gossypioides and G.
raimondii are sister species may be untenable.
Ano et al. (1982) nominated northeastern Brazil as a prob-able site for the origin of the allotetraploid lineage. Fryxell (1979) noted that all the wild populations of the tetraploid species occupy littoral or littoral-derived habitats. This obser-vation, in concert with a proposed divergence during the Plei-sotocene (Fryxell, 1965; Phillips, 1963; Wendel, 1989), a pe-riod of rapidly changing ocean levels and the fact that
Gos-sypium seeds are salt-water tolerant (Stephens, 1958; Fryxell,
1979), suggests that prevailing ocean currents would be a pri-mary determinant in the direction of migration of the nascent allopolyploids. The prevailing marine currents move from northeastern Brazil along the northeastern coast of South America passing through the Caribbean Sea into the Gulf of Mexico. Ano et al.’s (1982) hypothesis suggests that the al-lotetraploid lineage arose in northeastern Brazil and that sub-sequent northerly movement of germplasm with the prevailing marine currents along the coast of South America led to the colonization of northern coastal South America, Gulf coastal Mexico, and the Islands of the Antilles. Under this scenario,
G. mustelinum represents the resident descendent of the
na-scent allopolyploid, consistent with its basal position in gene topologies, while the colonial populations diverged into the other four allotetraploid species (see also Wendel, Rowley, and Stewart, 1994).
The convention that G. gossypioides and G. raimondii are sister species is based on gross comparative morphology and the interfertility of the two species (Brown and Menzel, 1952a, b). Although this conclusion is supported by phylogenetic analyses of chloroplast restriction site mutations (Wendel and Albert, 1992; Seelanan et al., 1997), it is not unassailable. In the first place, gross morphological comparisons are subject to contradictory interpretations. Although Brown and Menzel (1952a, b) concluded that G. gossypioides and G. raimondii are closely related, they also noted that G. gossypioides is intermediate between G. thurberi and G. raimondii in flower size, shape, and color, leaf shape, and stem texture, having ‘‘a marked superficial resemblance to the F1hybrid’’ between the
two (Brown and Menzel, 1952a, p. 120). Hutchinson, Silow, and Stephens (1947) interpreted the situation differently and classified G. gossypioides with G. thurberi and G. trilobum in section ‘‘Thurberana.’’ They did this on the basis that the sec-tion ‘‘Thurberana’’ species were glabrous (or mostly so) and had lobed leaves and entire or three-toothed epicalyx bracts, in contrast to the section ‘‘Klotzschiana’’ species, including G.
raimondii, which were characterized by pubescent leaves and
stems, entire leaves, and laciniate epicalyx bracts.
In contrast to the equivocal conclusions afforded by gross morphological comparisons, phylogenetic analyses of the 5S ribosomal DNA (Cronn et al., 1996) and the FAD2-1 (Fig. 1) intron both resolve G. gossypioides as the sole descendent of one lineage from the basal divergence in a monophyletic D-genome lineage. In the case of the 5S ribosomal DNA, a to-pology rejecting the basal position of G. gossypioides would require another six steps. Although the basal position of G.
gossypioides in the FAD2-1 topology is supported by decay
index of only 1, collapsing this branch still would not resolve
G. gossypioides and G. raimondii as sister taxa. Conversely,
the FAD2-1 topology resolves G. raimondii (decay index of 2) as basal to the D-subgenome monophyletic lineage. Al-though not all the D-genome diploid species are included in this analysis, G. gossypioides and G. raimondii are the only probable candidates (reviewed by Endrizzi, Turcotte, and Ko-hel [1985] and Wendel, Schnabel, and Seelanan [1995]). Con-sequently, the weight of the evidence promotes G. raimondii as the most likely extant taxon that is sister to the allotetraploid D-subgenome progenitor. Furthermore, Stephens (1944a, b), based on a genetic analysis of leaf shape alleles in Gossypium, concluded that genetic control of leaf shape in the allotetra-ploids most likely reflected the interaction of the leaf shape genes in the Old World A-subgenome progenitor in combi-nation with the leaf shape genes from a D-genome diploid with entire leaves, of which there are only four, viz., G. aridum, G.
armourianum, G. klotzschianum, and G. raimondii. Thus, it is
worthy of note that in the FAD2-1 topology, G. klotzschianum resolves as basal to the combined G. raimondii-allotetraploid D-subgenome lineage (Fig. 1).
A-genome-specific base pair change. This reasoning may also be applicable to the FAD2-1 intron, but given that it resides in a single copy gene, it is unlikely. Particularly notable are the 33 unambiguous base pair substitutions and indels the G.
gossypioides FAD2-1 intron shares with the other D-genome
taxa and that differentiate them from the Old World E- and A-genome lineages (Fig. 1). The G. gossypioides FAD2-1 intron contains none of the 13 A-genome-specific substitutions. Thus, chloroplast and nuclear genes place G. gossypioides in incon-gruent positions within the larger Gossypium topology. This incongruity can be explained by assuming that the chloroplast topology accurately tracks the taxon evolution and that the
FAD2-1, the 5S ribosomal repeat, and the ribosomal 18S–26S
DNA repeats are mosaics that have recombined with their A-genome orthologs. Alternatively, one can propose that the
FAD2-1 and 5S ribosomal genes accurately track the taxon
evolution. This implies that G. gossypioides obtained the G.
raimondii-like chloroplast via later interspecific introgression
rather than by inheritance. At the moment the weight of evi-dence favors the latter hypothesis. The key may lie in a better understanding of G. raimondii. The chloroplast DNA topology indicates that G. gossypioides and G. raimondii were in phys-ical contact at some point either via interspecific introgression or via a recent common ancestor. Understanding how G.
rai-mondii came to its present geographic isolation from its New
World congeners, particularly G. gossypioides, will be vital to finally resolving the incongruence in topological placement of
G. gossypioides in chloroplast and nuclear topologies.
Gossypium mustelinum is basal among the five allotetraploid species—The topological placement of both of
the G. mustelinum sequences is consistent with the growing consensus that it is the sole descendent of one branch of the earliest divergence within the allotetraploid lineage, the second lineage subsequently diverging into the other four allotetra-ploid species (Small et al., 1998). Within the robustly sup-ported D-subgenome lineage (decay index of 11), the G.
mus-telinum D-subgenome intron sequence lacks three
nonhomo-plasious substitutions that unite the remaining four species. The situation is more complex in the A-genome lineage. The A subgenome lacks defining synapomorphies and thus re-solves as paraphyletic to a monophyletic A-diploid lineage— probably reflecting the sluggish rate of base pair substitution in the A-genome lineage. Nonetheless, the G. mustelinum in-tron sequence also resolves as basal relative to the other A-subgenome sequences. Small et al. (1998) obtained an iden-tical result from analyses of the plastid trnT-trnL spacer region and nuclear alcohol dehydrogenase (AdhC) sequences.
Evolution of the Australian Gossypium species—Both the
distance- and parsimony-optimized topologies suggest that the original Gossypium entity in Australia very quickly diverged into four main lineages; the C genome, the K genome, G.
bickii, and G. australe/G. nelsonii (Fig. 1). Consistent with
previous phylogenetic analyses, the FAD2-1 intron topologies support the monophyly of the C- and K-genome lineages (Wendel and Albert, 1992; Seelanan, Schnabel, and Wendel, 1997; Seelanan et al., 1999). Two unambiguous substitutions and a decay index of 1 (Fig. 1) weakly support the C-genome monophyly. This resolution, however, is consistent with to-pologies derived from rDNA ITS (parsimony optimized) and
AdhD sequences (Seelanan, Schnabel, and Wendel, 1997;
See-lanan et al., 1999). Chloroplast restriction site topologies
re-solve G. robinsonii as basal, with G. sturtianum as basal to the remainder of the Australian species (Wendel and Albert, 1992), but when taxa with known reticulate histories are ex-cluded, G. robinsonii and G. sturtianum occur as unresolved basal lineages relative to the other Australian species in strict consensus trees (Seelanan, Schnabel, and Wendel, 1997). Thus, the chloroplast restriction data at least do not contradict the inferences based on the FAD2-1 intron and AdhD sequenc-es. The monophyly of the K genome is strongly supported and is consistent with all previous analyses and their distinctive suite of morphological features (Wendel and Albert, 1992; Seelanan, Schnabel, and Wendel, 1997; Seelanan et al., 1999). Based on the estimated age of the basal divergences, the age of the Australian Gossypium clade is probably 9.4–24.1 mil-lion years old (Table 4), a figure that coincides with the age of the earliest records of malvaceous pollen in Australia, es-timated to have been deposited 12–25 mybp (Muller, 1981, 1984).
One striking difference between the phylogenetic analysis of the FAD2-1 intron and previous cladistic analyses is the level of resolution and the estimated age of the earliest diver-gence within the K genome. Whereas topologies based on
rpl16, ITS, and AdhD sequences produced largely unresolved
rakes (Seelanan et al., 1999), the FAD2-1 identified two pri-mary sublineages (K1 and K2; Fig. 1), both of which are sup-ported by decay indices of 3. The K1 clade comprises plants with mostly upright habits and sporadic coastal or near-coastal distributions in contrast to the K2 clade, which contains spe-cies that are mostly prostrate and have more widespread pop-ulations inland (Fryxell, Craven, and Stewart, 1992). Estimates based on the FAD2-1 intron suggest that the first divergence among the K-genome species occurred 8.1–16.2 mybp (Table 4), a figure that stands in stark contrast to earlier estimates of 0.7–3 mybp (Seelanan et al., 1999).
The reticulate evolutionary history of G. bickii—Gossy-pium bickii is traditionally accepted as a member of a
mono-phyletic G-genome clade based primarily on morphological and cytogenetic similarities (Fryxell, 1979, 1992; Stewart, 1995), but recent sequence analysis of a portion of the AdhD gene (Seelanan et al., 1999) in which G. bickii appears as basal within the Australian Gossypium species undermines this in-terpretation. This incongruity is particularly interesting be-cause G. bickii is known to have arisen via or experienced a homoploid reticulate event involving the ancestor of G.
stur-tianum (Wendel, Stewart, and Rettig, 1991). The result is that G. bickii carries a G. sturtianum-like chloroplast, but retains a G. australe–G. nelsonii-like nuclear genome (Wendel and
Al-bert, 1992; Seelanan, Schnabel, and Wendel, 1997, Seelanan et al., 1999). The hybridization event that resulted in the trans-fer of the chloroplast would have brought the nuclear genomes into recombinational contact before the proto-G. sturtianum chromosomes were purged from the G. bickii genome, yet there is no evidence, to date, of any C-genome-derived nuclear sequences in the G. bickii genome. This raises the question of whether the G. bickii AdhD sequence represents an orphaned
G. sturtianum gene or a mosaic gene that combines features
of the original G. bickii gene with the G. sturtianum gene (cf. Cronn et al., 1996). If it does not, the incongruity between ITS-derived topologies that place G. bickii sister to G. australe and the AdhD topologies that place it basal to a well-defined
G. australe/G. nelsonii lineage may point to a second cryptic
The weight of the evidence favors the latter interpretation. The AdhD sequences place G. bickii basal to the G. australe/
G. nelsonii (decay index of 10) and the G. robinsonii/G. stur-tianum (decay index of 2) clades (Seelanan et al., 1999). The FAD2-1 intron topology also clearly places G. bickii as a basal
clade, again lacking the ten substitutions that unite G.
aus-trale–G. nelsonii into a single clade (decay index of 9; Fig. 1)
and the two base-pair substitutions that unite G. robinsonii and
G. sturtianum (decay index of 1; Fig. 1). Similarly, a
phylo-genetic analysis of allozyme alleles and rDNA 18S–26S re-striction site mutations placed G. bickii basal to a strongly supported G. australe-G. nelsonii clade (Wendel, Stewart, and Rettig, 1991), and in contrast to the observation that G.
aus-trale, G. bickii, and G. sturtianum have 15, 8, and 7
species-specific alleles, respectively, G. bickii shares only three alleles exclusively with G. australe and none with G. sturtianum. Furthermore, if the rDNA 18S–26S restriction site mutations specific to G. australe, G. bickii, and G. nelsonii are mapped onto the topology derived from the ITS sequences in Seelanan, Schnabel, and Wendel (1997) and Seelanan et al. (1999), the homoplasious loss of the KpnI restriction site in G. australe and G. bickii becomes a synapomorphy uniting the two, and the inferred gain of a BanI restriction site in the immediate ancestor of the G. australe/G. bickii/G. nelsonii clade followed by the immediate loss in the ancestor of G. australe/G.
nel-sonii becomes a single gain in G. bickii. Thus, of the four
nuclear sequences so far analyzed, three (isozyme alleles,
AdhD, and FAD2-1) consistently resolve G. bickii as a basal
lineage, and only the ITS sequences and restriction site dif-ferences suggest that G. bickii shares a more recent ancestor with G. australe than with any other Australian species.
It is highly improbable that this incongruity reflects the pres-ence of intact or mosaic G. sturtianum genes in the G. bickii nucleus. All chloroplast topologies are consistent in suggesting that the transfer of the proto-G. sturtianum chloroplast oc-curred after the divergence of G. robinsonii and G. sturtianum (Wendel and Albert, 1992; Seelanan, Schnabel, and Wendel, 1997; Seelanan et al., 1999). Thus, if these genomic sequences are intact residual genes from the G. sturtianum genome, they should resolve sister to G. sturtianum, not basal to a mono-phyletic G. sturtianum–G. robinsonii lineage as they do in the
AdhD and FAD2-1 topologies (Fig. 1; Wendel and Albert,
1992; Seelanan et al., 1999). Furthermore, of the 76 isozyme alleles (at 21 loci) surveyed by Wendel and Albert (1992), none are shared exclusively by G. bickii and G. sturtianum. The only remaining hypothesis is that the genes sampled to date are G. bickii–G. sturtianum mosaics, but it is unlikely that the AdhD, the FAD2-1, and the eight G. bickii-specific isozyme alleles are all products of intragenic recombination. Thus, based on the data currently at hand, the most probable explanation for the topological inconsistency of G. bickii is that it experienced two homoploid reticulate events, capturing its chloroplast genome from G. sturtianum and its ribosomal DNA repeat from G. australe. It is worthy of note, then, that recent field observations demonstrate that the biological means exist for introgression between G. australe and G. bickii. In 1997, two intermingled populations of G. australe and G.
bick-ii containing fertile hybrids were identified in the Australian
central arid zone (unpublished data). This indicates that intro-gression of the G. australe ribosomal DNA repeat could be recent and may not yet be fixed in G. bickii.
The inference that G. bickii represents a basal lineage among the Australian species may explain the morphological
singularities of G. bickii. In gross morphological appearance, particularly pubescence and flower color, it is similar to G.
australe and G. nelsonii, with whom G. bickii is classified
taxonomically in section Hibiscoidea (Fryxell, 1992). How-ever, G. bickii’s short stature and multistemmed habit contrast sharply with the taller less profusely branched habit of G.
aus-trale and G. nelsonii and is strongly reminiscent of the erect
and suberect habit of many of the K-genome species (Fryxell, 1979, 1992). Gossypium bickii also lacks the stiff spreading seed hairs of G. australe and G. nelsonii, instead possessing a seed vestiture identical to that of G. robinsonii and G.
stur-tianum.
Does the FAD2-1 intron enhance gene expression?—The
taxon topologies inferred from the phylogenetic analysis of the
FAD2-1 microsomal v-6 desaturase large intron sequences,
however useful and interesting in reconstructing historical evo-lutionary events in the genus Gossypium, are merely the first step toward understanding the regulatory role of introns in gene expression in cotton. A positive effect of introns on gene expression has been observed for many plant genes. Expres-sion of reporter genes under the control of the maize Adh1,
Sh1, Bx1, or Act promoter is increased up to several hundred
fold by the inclusion of an intron (Callis, Fromm, and Walbot, 1987; Maas et al., 1991; Oard, Paige, and Dvorak, 1989; Vasil et al., 1989). Similarly, Arabidopsis thaliana genes encoding polyubiquitin (Norris, Meyer, and Callis, 1993), transcription factor EF-1a (Currie et al., 1991, 1993) all have an intron in the 59 UTR region that increases the expression of reporter gene fusions 2.5- to 1000-fold relative to intron-less controls. This enhancement of gene expression has been ascribed to intron splicing (Gidekel, Jimenez, and Herrera-Estrella, 1997). In view of the remarkable conservation of the size and position of the 59 UTR intron of FAD2-1 across all the 31 Gossypium species examined and the presence of a 59UTR intron in other species, such as, Arabidopsis thaliana, it is possible that this intron might have an enhancing effect on the expression of
FAD2-1. Experiments are currently underway to examine
whether the 59 UTR intron on its own or in conjunction with the FAD2-1 promoter contributes to the regulation of expres-sion of FAD2-1 in cotton. These experiments will be all the more informative because they take place within a sound phy-logenetic framework.
LITERATURE CITED
ANO, G., J. SCHWENDIMAN, J. FERSING,ANDM. LACAPE. 1982. Les coton-niers primitifs G. hirsutum race yucatanense de al Pointe des Chaˆteaux en Guadeloupe et l’origine possible des cotonniers te´traploı¨des du Nou-veau Monde. Coton et Fibres Tropicales 37: 327–332.
BREMER, K. 1988. The limits of amino acid sequence data in angiosperm phylogenetic reconstruction. Evolution 42: 795–803.
BROWN, M. S.,ANDM. Y. MENZEL. 1952a. Additional evidence on the cross-ing behavior of Gossypium gossypioides. Bulletin of the Torrey Botanical Club 79: 285–292.
———,AND———. 1952b. The cytology and crossing behavior of Gos-sypium gossypioides. Bulletin of the Torrey Botanical Club 79: 110–125. BRUBAKER, C. L., F. M. BOURLAND,ANDJ. F. WENDEL. 1999. The origin and domestication of cotton. In C. W. Smith and J. T. Cothren [eds.], Cotton: origin, history, technology, and production. Wiley, New York, New York, USA.
5S ribosomal DNA in diploid and allopolyploid cottons. Journal of Mo-lecular Evolution 42: 685–705.
CURRIE, C., M. AXELOS, C. BARDET, R. ATANASSOVA, N. CHAUBET,ANDB. LESCURE. 1993. Molecular organisation and developmental activity of an Arabidopsis thaliana EF-1agene promoter. Molecular and General Genetics 218: 78–86.
——–, T. LIBOZ, C. BARDET, E. GANDER, C. ME´ DALE, M. AXELOS,ANDB. LESCURE. 1991. Cis- and trans-acting elements involved in the activa-tion of Arabidopsis thaliana A1 gene encoding the translaactiva-tion elongaactiva-tion factor EF-1a. Nucleic Acids Research 19: 1305–1310.
DONOGHUE, M. J., R. G. OLMSTEAD, J. F. SMITH,ANDJ. D. PALMER. 1992. Phylogenetic relationships of Dipsacales based on rbcL sequences. An-nals of Missouri Botanical Garden 79: 333–345.
ENDRIZZI, J. E., E. L. TURCOTTE,ANDR. J. KOHEL. 1985. Genetics, cytology, and evolution of Gossypium. Advances in Agronomy 23: 271–375. ERIKSSON, T.,ANDN. WIKSTRO¨ M. 1995. Autodecay ver. 3.0 (program
dis-tributed by the authors). Dept. of Botany, Stockholm University, Stock-holm, Sweden.
FRYXELL, P. A. 1965. Stages in the evolution of Gossypium L. Advancing Frontiers of Plant Sciences 10: 31–55.
———. 1979. The natural history of the cotton tribe (Malvaceae, Tribe Gos-sypieae). Texas A&M Press, College Station, Texas, USA.
———. 1992. A revised taxonomic interpretation of Gossypium L. (Mal-vaceae). Rheedea 2: 108–165.
——–, L. A. CRAVEN,ANDJ. MCD. STEWART. 1992. A revision of Gossy-pium sect. Grandicalyx (Malvaceae), including the description of six new species. Systematic Botany 17: 91–114.
GIDEKEL, M., B. JIMENEZ,ANDL. HERRERA-ESTRELLA. 1997. The first in-tron of the Arabidopsis thaliana gene coding for elongation factor 1b contains an enhancer-like element. Gene 170: 201–206.
HUTCHINSON, J. B., R. A. SILOW,ANDS. G. STEPHENS. 1947. The evolution of Gossypium and the differentiation of the cultivated cottons. Geoffrey Cumberlege/Oxford University Press, London, UK.
JUKES, T. H.,ANDC. R. CANTOR. 1969. Evolution of protein molecules. In H. N. Munro [ed.], Mammalian protein metabolism, 21–132. Academic Press, New York, New York, USA.
KUMAR, S., K. TAMURA,ANDM. NEI. 1993. MEGA: Molecular evolutionary genetic analysis, version 1.01. Pennsylvania State University, University Park, Pennsylvania, USA.
LIU, Q., S. P. SINGH, C. L. BRUBAKER, P. J. SHARP, A. G. GREEN,ANDD. R. MARSHALL. 1996. Isolation and characterisation of two different micro-somalv-6 desaturase genes in cotton (Gossypium hirsutum L.). Pro-ceedings of the 12th International Symposium on Plant Lipids, 7–12 July, 1996, Toronto, Canada. Kluwer Academic Publishers, Dordrecht, The Netherlands.
———, ———, ———, ———, ———,AND———. 1997. Character-ization of a large intron in 59UTR of microsomalv-6 desaturase gene from Gossypium spp. and other plant species. Abstracts of the Fifth In-ternational Congress of Plant Molecular Biology, Singapore. 21–27 Sep-tember, 1997, Singapore. Plant Molecular Biology Reporter 15(3 sup-plement), Abstract.
———, ———, ———, ———, ———,AND———. 1999. Molecular cloning and expression of a cDNA encoding a microsomalv-6 fatty acid desaturase from cotton (Gossypium hirsutum). Australian Journal of Plant Physiology: 26: 101–106.
MAAS, C., J. LAUFS, S. GRANT, C. KORFHAGE,ANDW. WERR. 1991. The combination of a novel stimulatory element in the first exon of the maize shrunken-1 gene with the following intron enhances reporter gene ex-pression up to 1000-fold. Plant Molecular Biology 16: 199–207. MULLER, J. 1981. Fossil pollen records of extant angiosperms. Botanical
Review 47: 1–142.
———. 1984. Significance of fossil pollen for angiosperm history. Annals of the Missouri Botanical Garden 71: 419–443.
NORRIS, S. R., S. E. MEYER,ANDJ. CALLIS. 1993. The intron of Arabidopsis thaliana polyubiquitin genes is conserved in location and is a quantitative determinant of chimeric gene expression. Plant Molecular Biology 21: 895–906.
OARD, J. H., D. PAIGE, ANDJ. DVORAK. 1989. Chimeric gene expression using maize intron in cultured cells of breadwheat. Plant Cell Reports 8: 156–160.
OKULEY, J., J. LIGHTNER, K. FELDMANN, N. YADAV, E. LARK, AND J. BROWSE. 1994. Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid synthesis. Plant Cell 6: 147–158. PATERSON, A. H., C. L. BRUBAKER,ANDJ. F. WENDEL. 1993. A rapid
meth-od for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Molecular Biology Reporter 11: 122–127. PHILLIPS, L. L. 1963. The cytogenetics of Gossypium and the origin of New
World cottons. Evolution 17: 460–469.
SAMBROOK, J., E. F. FRITSCH,ANDT. MANIATIS. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, New York, New York, USA.
SEELANAN, T., C. L. BRUBAKER, J. MCD. STEWART, L. A. CRAVEN,ANDJ. F. WENDEL. 1999. Molecular systematics of Australian Gossypium sec-tion Grandicalyx (Malvaceae). Systematic Botany 24: 183–208. ———, A. SCHNABEL,ANDJ. F. WENDEL. 1997. Congruence and consensus
in the cotton tribe (Malvaceae). Systematic Botany 22: 259–290. SHANKLIN, J.,ANDE. B. CAHOON. 1998. Desaturation and related
modifi-cations of fatty acids. Annual Review of Plant Physiology and Plant Molecular Biology 49: 611–641.
SIMPSON, G. G.,ANDW. FILIPOWICZ. 1996. Splicing of precursors to mRNA in higher plants: mechanism, regulation and sub-nuclear organisation of the spliceosomal machinery. Plant Molecular Biology 32: 1–41. SMALL, R. L., J. A. RYBURN, R. C. CRONN, T. SEELANAN,ANDJ. F. WENDEL.
1998. The tortoise and the hare: choosing between noncoding plastome and nuclear ADH sequences for phylogeny reconstruction in a recently diverged plant group. American Journal of Botany 85: 1301–1315. STEPHENS, S. G. 1944a. The genetic organization of leaf-shape development
in the genus Gossypium. Journal of Genetics 46: 28–51.
———. 1944b. Phenogenetic evidence for the amphidiploid origin of new world cottons. Nature 153: 53–54.
———. 1958. Salt water tolerance of seeds of Gossypium species as a pos-sible factor in seed dispersal. American Naturalist 92: 83–92.
STEWART, J. MCD. 1995. Potential for crop improvement with exotic germ-plasm and genetic engineering. In G. A. Constable and N. W. Forrester [eds.], Challenging the future: Proceedings of the World Cotton Research Conference-1, Brisbane Australia, February 14–17, 313–327. CSIRO, Melbourne, Australia.
———, L. A. CRAVEN,ANDJ. F. WENDEL. 1997. A new Australian species of Gossypium. In Proceedings of the Beltwide Cotton Conferences 448. National Cotton Council, Memphis, Tennessee, USA.
SWOFFORD, D. L. 1991. PAUP: Phylogenetic analysis using parsimony, ver-sion 3.1. Illinois Natural History Survey, Champaign, Illinois, USA. TAJIMA, F. 1993. Simple methods for testing the molecular evolutionary
clock hypothesis. Genetics 135: 599–607.
———,AND M. NEI. 1984. Estimation of evolutionary distance between nucleotide sequences. Molecular Biology and Evolution 1: 269–285. VASIL, V., M. CLANCY, R. J. FERL,ANDI. K. VASIL. 1989. Increased gene
expression by the first intron of maize shrunken-1 locus in grass species. Plant Physiology 91: 1575–1579.
WENDEL, J. F. 1989. New world tetraploid cottons contain old world cyto-plasm. Proceedings of the National Academy of Sciences, USA 86: 4132– 4136.
———ANDV. A. ALBERT. 1992. Phylogenetics of the cotton genus (Gos-sypium): character-state weighted parsimony analysis of chloroplast-DNA restriction site data and its systematic and biogeographic implica-tions. Systematic Botany 17: 115–143.
———, R. ROWLEY,ANDJ. MCD. STEWART. 1994. Genetic diversity in and phylogenetic relationships of the Brazilian endemic cotton, Gossypium mustelinum (Malvaceae). Plant Systematics and Evolution 192: 49–59. ———, A. SCHNABEL,ANDT. SEELANAN. 1995. An unusual ribosomal DNA
sequence from Gossypium gossypioides reveals ancient, cryptic, inter-genomic introgression. Molecular Phylogenetics and Evolution 4: 298– 313.
———, J. MCD. STEWART,ANDJ. H. RETTIG. 1991. Molecular evidence for homoploid reticulate evolution among Australian species of Gossy-pium. Evolution 45: 694–711.