Disease
Jennifer Court, Carmen Martin-Ruiz, Margaret Piggott, Dean Spurden,
Martin Griffiths, and Elaine Perry
Loss of cortical nicotinic acetylcholine receptors with high affinity for agonists (20 –50%) in patients with Alzhei-mer’s disease is a common finding. Recent immunochemi-cal analyses indicate that this deficit is predominantly associated with the loss of a4 subunits (30 –50%),
al-though modest reductions of a3 may occur in some
individuals (25–29%). No reduction ofb2 subunit protein expression or levels of a3 and a4 messenger RNA has been reported. Decline in cortical [125I]a-bungarotoxin binding anda7 protein expression does not appear to be as extensive or widespread as the loss of a4 (0 – 40%), with no reduction in messenger RNA expression. In the thalamus, there was a trend for reduced [3H]nicotine binding in the majority of nuclei (0 –20%) in Alzheimer’s disease; however, there was a significant decline in [125I]a-bungarotoxin binding in the reticular nucleus. In the striatum [3H]nicotine binding was reduced in Alzhei-mer’s disease, and although neuroleptic medication ac-centuated this change, it occurred in those free of neuro-leptics. Changes in nicotinic acetylcholine receptors in Alzheimer’s disease are distinct from those in normal aging and are likely to contribute to clinical features and
possibly neuropathology. Biol Psychiatry 2001;49:
175–184 © 2001 Society of Biological Psychiatry
Key Words: Nicotinic acetylcholine receptors,
Alzhei-mer’s disease, protein expression, cerebral cortex, thala-mus, striatum
Introduction
I
dentification of the loss of cholinergic neurons in the basal forebrain and of cholinergic innervation of the cerebral cortex in Alzheimer’s disease (AD) (for reviews, see Bowen 1983; Cummings and Benson 1987; Perry et al 1994) was followed by investigations into the involvementof cholinergic receptors. In contrast to choline acetyltrans-ferase, no major or consistent changes in muscarinic acetylcholine receptors (mAChRs) were observed in the cerebral cortex (for a review, see Nordberg 1992), al-though moderate reductions in the cortical M2 receptor subtype have been reported (Flynn et al 1995). In contrast, reductions in nicotinic acetylcholine receptors (nAChRs) (measured using radiolabeled nicotinic agonists such as [3H]acetylcholine, [3H]nicotine, and [3H]methyl carbamyl choline at nmol/L concentrations) ranging between 20% and 50% were consistently observed at autopsy in a number of neocortical areas and hippocampi of patients with AD (for reviews, see Court and Perry 1995; Kellar and Wonnacott 1990; Nordberg 1992). A significant reduction in [125I]a-bungarotoxin binding to a separate subtype of nAChRs was also reported in the temporal but not the frontal cortex in AD (Davies and Feisullin 1981; Sugaya et al 1990). The functional significance of this attenuation of brain nAChRs in AD was intriguing, partly as the density of nAChRs in the normal adult human neocortex was low relative to mAChRs (around a 40-fold difference in density; e.g., Court et al 1997) and also since the precise role(s) of nAChRs in endogenous cholinergic neurotransmission are unclear (Clarke 1993, 1995).
Extensive research over the last decade has established that brain nAChRs are a structurally and functionally diverse group of ligand-gated cation channels that are associated with numerous transmitter systems for which they have a modulatory function (Lindstrom et al 1995; McGehee et al 1995; McGehee and Role 1995; Wonnacott 1997). The majority of receptors with a high affinity for agonists (nicotine, methyl carbamyl choline, cytisine, and epibatidine) are composed of a4 and b2 subunits, but other subunits (e.g.,a3 anda6) may play a role in specific brain pathways (Clarke and Reuben 1996; Le Novere et al 1996) and modify receptor pharmacology (McGehee and Role 1995).a-Bungarotoxin (aBGT) binds to homomeric a7-containing receptors, which are characterized by a low sensitivity to agonists (Clarke 1992), a relatively high Ca21 conductance (McGehee and Role 1995), and a central nervous system distribution distinct from nAChRs with high affinity for agonists in the human brain (Figure
From the Joint MRC Newcastle University Centre Development in Clinical Brain Aging, Institute for the Health of the Elderly, Newcastle General Hospital, Newcastle upon Tyne, United Kingdom.
Address reprint requests to Jennifer Court, Newcastle General Hospital, Joint MRC Newcastle University Centre Development in Clinical Brain Aging, Institute for the Health of the Elderly, MRC Building, Newcastle upon Tyne NE4 6BE, United Kingdom.
Received May 22, 2000; revised November 27, 2000; accepted December 4, 2000.
© 2001 Society of Biological Psychiatry 0006-3223/01/$20.00
1) (Court and Perry 1995). Since nAChR subtypes may represent therapeutic targets with some level of pathway specificity, and changes in their expression could reflect disease specific pathology, there is currently great interest in evaluating nAChR subtype and subunit changes in AD.
Nicotinic Subtype and Subunit Changes in
Cortical Areas
Receptors with High Affinity for Agonists
Early reports that nAChRs with high affinity for agonists were reduced in neocortical areas relative to age-matched control subjects have been confirmed by more recent studies. These include positron emission tomography (PET) analyses employing (S)(2)[11C]nicotine (Nordberg et al 1990, 1995), which demonstrated nicotine-binding deficits in the temporal and frontal cortex in AD in vivo, although this technique does not control for nonspecific or endogenous binding and a component of the [11C]nicotine signal may reflect cerebral blood flow. This attenuation in nicotinic agonist binding could reflect deficits in both cortical innervation and intrinsic neurons: messenger RNAs (mRNAs) for botha3 anda4 subunits have been reported to be present in cortical neurons (Wevers et al 1994), and experimental lesions in rats indicate the pres-ence of nAChRs with high affinity for agonists associated with thalamocortical pathways (Prusky et al 1987; Sahin et al 1992), although their presence on cortical cholinergic inputs is less certain (Miyai et al 1990; Tilson et al 1989; Wenk and Rokaeus 1988).
Autoradiographic analysis of [3H]nicotinic binding in the human hippocampus and adjacent cortex indicates the highest densities in the subicular complex, stratum lacu-nosum moleculare, dentate gyrus, and entorhinal cortex (Perry et al 1992; Court et al, previously unpublished data [Figure 1]). This pattern of binding suggests an association with the perforant pathway, although results of a lesion study in rats would tend to refute this (Aubert et al 1994). In addition, functional studies of rat tissue indicate nAChRs with high affinity for agonists may have a number of functional roles within the hippocampus—for example, modulation of noradrenaline, acetylcholine, and g-aminobutyric acid (GABA) release (Alkondon et al 1999; Clarke and Reuben 1996; Wilkie et al 1996). Reductions in [3H]nicotinic binding in AD of around 30% relative to age-matched elderly control subjects extend throughout the hippocampal formation (Perry et al 1995) (Table 1). This generalized loss in AD would argue against the reduction in nAChRs being linked with a single pathway within the hippocampus.
The radioligands [3H]cytisine, [3H]ABT 418, and
[3H]epibatidine have allowed a level of selective evalua-tion of the nAChR loss in AD. Cytisine and ABT 418 are considered to bind predominantly to a4/b2 subunits, whereas epibatidine may bind in addition to a3 and possibly other subunit-containing receptors (Flores et al 1992, 1997; Warpman and Nordberg 1995; Zoli et al 1998). By comparison of [3H]ABT 418 and [3 H]epibati-dine binding it has been adduced that it is primarily the former subtype that is lost from the cerebral cortex in AD
Figure 1. [3
H]Nicotine (A, C) and [125
I]a -bungarotoxin binding (B, D) in the human hippocampal formation (A, B) and thala-mus (C, D) at the level of the lateral geniculate nucleus. Autoradiography was according to Court et al (1997) and Spurden et al (1997) using 1.2 nmol/L [125
I]a -bun-garotoxin and 4 nmol/L [3
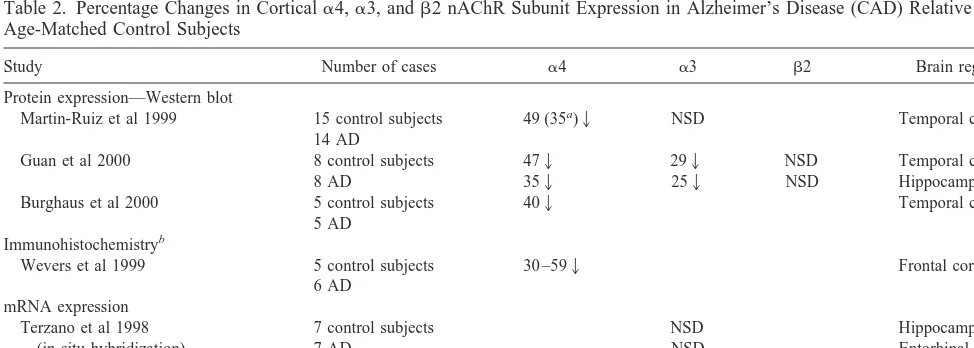
(Warpman and Nordberg 1995). Nicotinic acetylcholine receptor subunit deficits are now being actively explored immunochemically. A consistent pattern emerges from a number of published reports (Burghaus et al 2000; Guan et al 2000; Martin-Ruiz et al 1999; Wevers et al 1999) in relation toa4 subunit protein expression of a reduction in AD in both neocortical areas and the hippocampus (Table 2). This reduction in the a4 subunit was shown in one study to be proportional to the loss in epibatidine binding measured in immediately adjacent sections of cortex in the same cases (Martin-Ruiz et al 1999). In contrast, a recent study has indicated no reduction inb2 subunits (Guan et al 2000). A small but statistically significant reduction in the
a3 subunit was demonstrated in both the temporal cortex and the hippocampus by Guan et al (2000), but not by Martin-Ruiz et al (1999) in the temporal cortex. These data indicate that the major contributor to the loss of high-affinity nAChRs in cortical areas in AD is likely to be the a4 subunit, although this does not preclude a minor contribution from the loss of other subunits. The reduction ina4 subunit protein does not appear to be the result of attenuated gene transcription because mRNA levels in AD appear comparable to those of age-matched control sub-jects (Hellstrom-Lindahl et al 1999).
a7-Containing Receptors
With regard toa7 nAChR subunit protein expression, the extent of deficits in cerebral cortical areas in AD may be more restricted than fora4-containing receptors in terms of magnitude, areas involved, and consistency between different groups of cases. No significant difference in [125I]aBGT binding in the frontal cortex was observed in
two independent studies (Davies and Feisullin 1981; Sugaya et al 1990), and although the early study of Davies and Feisullin reported a 40% reduction in the temporal cortex, this was not replicated by a more recent analysis (Hellstrom-Lindahl et al 1999). This lack of a consistent deficit in corticalaBGT binding in AD cases is perhaps surprising because lesions of the basal forebrain in rats suggest that these receptors are at least partly present on cholinergic terminals (Sugaya et al 1991).
In the hippocampus a 25% reduction in [125I]aBGT binding (using membrane homogenates) was observed in AD relative to age-matched control subjects, although the number of control cases studied was small (n 5 4) Table 1. [3
H]Nicotine Binding in the Hippocampus
Subfields Control Alzheimer
CA1 9.3864.08 5.9861.62a
CA2/3 7.2763.94 6.2562.18
CA4 6.3263.03 5.4562.58
Stratum lacunosum 11.8163.99 8.6863.29a
Dentate gyrus 10.0464.41 7.5362.11
Subiculum 13.1665.26 10.1763.53
Entorhinal cortex 1 10.0864.92 7.1662.40
Entorhinal cortex 2 11.0564.95 7.7762.49a
Entorhinal cortex 3 14.4567.25 10.4363.49
Entorhinal cortex 4 9.5264.95 6.9263.33
Perirhinal cortex 12.4366.32 9.2763.55
Analysis was of 14 control subjects and 14 Alzheimer’s disease (AD) cases, mean ages 7769 and 8065 years and postmortem delays 29614 and 30617 hours, respectively. Values are means6SDs determined using 4 nmol/L [3
H]ni-cotine according to Court et al (1997). For layers 1– 4 in the entorhinal cortex, see Figure 1A.
aIndicates significant difference (p,.05, two-sample independent t test).
Overall there was a highly significant trend for reduced [3H]nicotine binding in AD
cases relative to control subjects [two-way analysis of variance, F(1,270)56.47,
p5.000].
Table 2. Percentage Changes in Corticala4,a3, andb2 nAChR Subunit Expression in Alzheimer’s Disease (CAD) Relative to Age-Matched Control Subjects
Study Number of cases a4 a3 b2 Brain region
Protein expression—Western blot
Martin-Ruiz et al 1999 15 control subjects 14 AD
49 (35a)2 NSD Temporal cortex
Guan et al 2000 8 control subjects
8 AD
Burghaus et al 2000 5 control subjects
5 AD
402 Temporal cortex
Immunohistochemistryb
Wevers et al 1999 5 control subjects
6 AD
30 –592 Frontal cortex
mRNA expression
NSD, no statistically significant difference; mRNA, messenger RNA; RT-PCR, quantitative reverse transcription polymerase chain reaction. aDecrease relative to known nonsmoking control subjects.
(Hellstrom-Lindahl et al 1999). In a larger autoradio-graphic analysis of [125I]aBGT binding in the hippocam-pal formation and entorhinal cortex (n 5 14 in AD and control groups), considerable variation between cases, particularly AD ones, was observed, and there were no consistent or statistically significant differences between groups in any subarea (previously unpublished data) (Ta-ble 3).
Reports of Western blot analyses using antibodies directed against a7 subunit sequences also suggest no significant or only minor reductions in the temporal cortex in AD (Burghaus et al 2000; Guan et al 2000; Martin-Ruiz et al 1999) (Table 4). In the one study of a7 protein expression in the hippocampus a 36% reduction in AD cases relative to control subjects was reported (Guan et al 2000), and an immunochemical study also suggested a reduction in the number of cells expressinga7 protein in the frontal cortex in AD (Wevers et al 1999). In terms of mRNA expression of thea7 subunit, no difference was observed between AD and control subjects in the temporal
cortex, but interestingly a 65% increase was observed in the hippocampus (Hellstrom-Lindahl et al 1999). The latter finding might indicate that a compensation mecha-nism involving increased transcription of thea7 gene in the hippocampus occurs in a proportion of patients with AD. The potential importance of this finding, together with the relatively small numbers of cases, warrants further investigation.
Subcortical Areas
Nicotinic Receptors in the Thalamus in AD
The thalamus is a brain area of particularly intense expression of nAChRs. The density of nicotinic agonist binding in the human thalamus is high in the majority of nuclei, most strikingly in the lateral geniculate, medial geniculate, and anterior (Figure 1 and Table 5) (Breese et al 1997b; Spurden et al 1997; Xuereb et al 1990).a3 and, to a lesser extent, b2 subunit mRNAs are expressed in thalamic nuclei in the human thalamus (Rubboli et al 1994) and high levels of expression of a3, a4, and b2 mRNAs occur in the rodent thalamus (Marks et al 1992; Wada et al 1989), suggesting that nAChRs with high affinity for agonists are at least in part present on intrinsic neurons within this brain structure. In contrast to high-affinity agonist binding, [125I]aBGT binding and a7 mRNA are only highly expressed in the reticular nucleus of the thalamus, with considerably lower densities in other nuclei (Figure 1 and Table 6) (Breese et al 1997a; Spurden et al 1997).
Recent, novel, autoradiographic analysis of [3 H]nico-tine binding (Table 5) confirms an earlier report of no major deficits in AD patients relative to age-matched Table 3. [125
I]a-Bungarotoxin Binding in the Hippocampus
Subfields Control Alzheimer
CA1 3.4362.19 2.9461.94
CA2/3 2.2361.58 3.5262.78
CA4 2.6062.06 2.8862.02
Dentate gyrus 2.2761.68 3.4662.33
Subiculum 2.7861.25 2.6261.66
Perirhinal cortex 2.7861.89 2.3462.21
Entorhinal cortex 3.1162.05 3.3562.21
Analysis was of 14 control subjects and 14 Alzheimer’s disease (AD) cases, mean ages 7769 and 8065 years and postmortem delay, 29614 and 30617 hours, respectively. Values are means 6 SDs determined using 1.2 nmol/L [125I]a-bungarotoxin according to Court et al (1997). No significant difference was
found between AD and control subjects.
Table 4. Percentage Changes ina7 Nicotinic Acetylcholine Receptors in Alzheimer’s Disease (AD) Relative to Age-Matched Control Subjects
Study Number of cases Change Brain area
Protein expression by Western blot
Martin-Ruiz et al 1999 15 control subjects 14 AD
NSD Temporal cortex
Guan et al 2000 8 control subjects
8 AD
NSD 362
Temporal cortex Hippocampus
Burghaus et al 2000 5 control subjects
5 AD
162 Temporal gyrus
Immunohistochemistrya
Wevers et al 1999 5 control subjects
6 AD
20 – 622 Frontal cortex
mRNA by quantitative RT-PCR
Hellstrom-Lindahl et al 1999 5– 6 NSD
NSD 651
Temporal cortex Cerebellum Hippocampus
NSD, no statistically significant difference; mRNA, messenger RNA; RT-PCR, quantitative reverse transcription polymerase chain reaction.
aThe measure applied was the ratio of the number of cells expressinga7 immunoreactivity/the number of cells expressinga7
control subjects (Xuereb et al 1990); however, there was a trend for [3H]nicotine binding to be lower in the majority of nuclei in AD (Table 5) [two-way analysis of variance of density in both groups and all areas indicated a significant difference between AD and control cases; F(1,55)59.21,
p5.003]. For [125I]aBGT binding there was a significant reduction of 34% in the overall density in the reticular nucleus in AD (p5.021, independent t test) and a greater reduction (50%, p, .001) in islands of cells within the reticular nucleus, which showed more intense binding (Figure 1 and Table 6). This loss of [125I]aBGT binding is
not apparently specific for AD, since a similar reduction was observed in dementia with Lewy bodies and schizo-phrenia (Court et al 1999), which raises issues of its etiopathologic and clinical significance. The reticular nu-cleus receives input from the basal forebrain (Chen and Bentivoglio 1993), so that reduced [125I]aBGT binding may reflect presynaptic sites on attenuated cholinergic afferents. Interestingly, this nucleus is also particularly susceptible to cortical hypoxia (Hossmann 1999), possibly via uncontrolled glutamate release from numerous cortical inputs. Reduced [125I]aBGT binding in AD could
there-fore also reflect a loss of postsynaptic receptors on neurons within the reticular nucleus as a consequence of attrition of cortical glutamatergic neurons and disruption of energy metabolism in AD (Minoshima et al 1999; Rapoport 1999). Reduced facilitation of GABAergic neu-rons within this nucleus, which is involved in synchroniz-ing thalamic activity, may contribute to the noncognitive deficits (e.g., those in sensory processing giving rise to hallucinations) observed in AD. There was no significant change in [125I]aBGT binding in any other thalamic nuclei (Table 6).
Nicotinic Receptors in the Striatum in AD
Some previous autopsy studies, but not all, indicate reductions in high-affinity nicotinic agonist binding in the striatum (particularly in the caudate nucleus) in AD (Aubert et al 1992; Perry et al 1989; Rinne et al 1991; Shimohama et al 1986). These deficits, present in at least some AD patients, are likely to be associated with disor-ders of movement and mood observed in late stages of AD. However, a proportion of patients will have received long-term medication for the treatment of noncognitive symptoms, most notably with traditional neuroleptics. These drugs have a major effect on nigrostriatal dopami-nergic neurons, on which a high proportion of nAChRs are believed to be sited within the striatum (Clarke and Pert 1985). In a recent study from our laboratory (Court et al 2000) patients with AD taking neuroleptics tended to have greater loss of striatal [3H]nicotine binding than those who had not, although this was not as marked as in patients with Lewy body dementia (who suffer moderate loss of substantia nigra dopaminergic neurons) (Figure 2), and there was still a significant reduction of striatal [3 H]nico-tine binding in AD patients free of neuroleptics relative to age-matched control subjects.
In Parkinson’s disease the loss of striatal [3H]nicotine binding by 40 –70% closely parallels the loss of nigrostri-atal dopaminergic markers; however, this is not the case in AD (Table 7), suggesting a different mechanism occurring in AD leading to striatal nAChR loss. In AD, loss of nAChRs possibly situated on striatal intrinsic GABAergic Table 5. [3
H]Nicotine Binding in the Thalamus
Nuclei Control (N) Alzheimer (N)
Anterior 28.7267.49 (7) 23.8765.38 (13)
Ventral anterior 18.5564.38 (6) 14.7264.92 (6) Ventral lateral 17.8368.33 (6) 16.5064.94 (11)
Mediodorsal 23.3164.71 (9) 23.8064.80 (11)
Parafascicular 10.8263.36 (4) 8.9464.10 (5)
Centromedian 3.6263.45 (3) 4.6764.32 (4)
Ventroposteriormedial 17.7062.58 (3) 15.8965.39 (4) Ventroposteriorlateral 12.0863.8 (8) 10.3564.31 (5) Lateral dorsal 23.3661.53 (4) 19.4462.60 (4) Lateral posterior 20.1766.60 (6) 16.4665.13 (5)
Pulvinar 20.4563.60 (7) 17.3765.85 (7)
Reticular 11.9462.90 (11) 12.8463.99 (12)
Lateral geniculate 31.6565.93 (6) 26.1765.86 (6) Medial geniculate 29.5466.02 (4) 25.0264.32 (6)
Results are means6SDs. Binding was measured using 4 nmol/L [3H]nicotine
according to Spurden et al (1997). Eleven control and 13 Alzheimer’s disease (AD) cases were used for the analysis, mean ages 76614 and 8069 years and postmortem delays 28611 and 31615 hours, respectively. Tissue for all nuclei was not available for all cases. Although there was no significant difference in binding for any nucleus between AD and control cases, two-way analysis of variance indicated a significant overall trend for reduced [3H]nicotine binding in
AD [F(1,155)59.21, p5.003]. N, number of determinations.
Table 6. [125
I]a-Bungarotoxin Binding in the Thalamus
Nuclei Control (N) Alzheimer (N)
Anterior 1.0060.77 (6) 1.1460.84 (13)
Ventral anterior 1.4461.18 (4) 1.1461.25 (8) Ventral lateral 1.4160.65 (7) 1.9161.13 (7)
Mediodorsal 1.3560.38 (6) 1.8461.04 (9)
Centromedian 1.6661.32 (4) 1.9760.92 (5)
Ventroposteriormedial 1.3760.96 (5) 1.9060.35 (5) Ventroposteriorlateral 1.6160.38 (3) 1.6460.71 (8) Lateral posterior 1.3561.09 (5) 1.6760.71 (8)
Pulvinar 1.2260.68 (7) 1.4261.17 (4)
Reticular (overall) 6.2961.84 (11) 4.1662.31 (13)a Reticular (areas of
particularly high intensity)
14.1064.03 (10) 7.2063.58 (13)a
Lateral geniculate 2.1761.24 (5) 2.2961.51 (7) Medial geniculate 1.0361.02 (4) 2.0061.28 (4)
Results are means6SDs. Binding was measured using 1.2 nmol/L [125
I]a-bungarotoxin according to Spurden et al (1997). Eleven control and 13 Alzheimer’s disease (AD) cases were used for the analysis, mean ages 76614 and 8069 years and postmortem delays 28611 and 31615 hours, respectively. Tissue for all nuclei were not available for all cases. N, number of determinations.
aSignificant difference between AD and control values (p,.05, two-sample
or cholinergic neurons or glutamatergic or serotonergic afferents may occur.
Relationship to Normal Aging
In some respects, reductions in nAChRs in AD, particu-larly of those with high affinity for agonists, may be viewed as an accentuation of the normal process of aging. Reductions in these receptors in cortical areas with ad-vancing age in humans have been reported by many
groups, also in the striatum after the age of 70 years but not in the thalamus (Hellstrom-Lindahl and Court, in press; Nordberg 1993). However, more detailed analysis of nAChR expression indicates that patterns of change with age are not consistently reflected in AD. In the different subfields of the hippocampal formation the rate of change during senescence varies—for example, being more striking in the entorhinal cortex than the CA1 (Court et al 1997)— but in AD these areas display a similar magnitude of decline (Table 1). There is also evidence to suggest that the significant reductions in a3 and b2 subunit mRNA expression in the human cerebral cortex with age (Terzano et al 1998; Tohgi et al 1998) are not reflected in AD (Table 2). In addition, the reduction in [125I]aBGT binding in the entorhinal cortex observed with normal aging (Court et al 1997) was not paralleled by reduced [125I]aBGT binding in this area in AD (Table 3). These results indicate that the mechanisms involved in age-related nAChR loss may not be similar to those occurring in AD, but rather that changes taking place during normal aging may be one of the factors that predispose patients to the specific deficits that develop in AD.
Relationship of nAChR Loss to Alzheimer’s
Pathology
A key question is whether changes in nAChRs in AD are simply a reflection of neuronal pathology or are a signif-icant contributing factor to the etiology of AD. There is no doubt that attenuation of nAChR binding occurs in areas of notable pathology in AD, and a number of studies Figure 2. [3
H]Nicotine binding and dopamine levels in the caudate nucleus in Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB), derived from Court et al (in press). Subgroups of patients taking neuroleptic medications and those free of neuroleptics are indicated by1nl and2nl, respectively. Bars are means6SDs, and the figures on bars the number of cases. All means are significantly different from control subjects (p , .05). *Subgroups of DLB patients (2nl vs. 1nl) were significantly different (p,.05).
Table 7. Basal Ganglia Changes in Alzheimer’s (AD) and Parkinson’s (PD) Diseases
Control Alzheimer Parkinson
SN pars compacta neuronsa 4946142 4896197 156682b
Dopaminea
Caudate N s50%
Putamen N s80%
HVA/DAc
Caudate N a300%
Putamen N a1,000%
[3H]nicotine binding (fmol/mg tissue
equivalent)d
Ventral caudate 9.0163.21 5.0661.64b 5.1062.95b
Dorsal putamen 8.4463.06 6.2661.26b 2.7461.88b
ChATaef
Striatum sN N
Nicotine binding: control subjects N542, AD N513, PD N513. Arrows indicate differences relative to control subjects. SN, substantia nigra; HVA/DA, homovanillic acid/dopamine; ChAT, choline acetyltransferase.
aData derived from Perry et al (1998).
bSignificant difference from control subjects (p,.05).
cData derived from Piggott et al (1998).
dData derived from Court et al (2000).
eData derived from Aubert et al (1992).
indicate a significant correlation between AD pathology and nAChR loss. Even in the striatum, where disease-related changes in dopamine parameters do not occur as in Parkinson’s disease (Table 7), pathology in terms of amyloid accumulation and loss of choline acetyltrans-ferase has been reported (Gearing et al 1997; Perry et al 1998; Shimohama et al 1986). There appears to be a relationship between nicotinic receptor density and cogni-tive function: in both in vivo and autopsy studies the intensity of high-affinity nicotinic agonist binding in the temporal cortex has been significantly correlated to de-mentia rating (Nordberg et al 1995; Perry et al 2000). Further, treatment of AD patients with an acetylcholines-terase inhibitor was associated with both an improvement of cognitive performance and an increase in nicotine binding as measured by PET (Nordberg 1999). In relation to the loss of cortical synapses in AD, [3H]epibatidine has been shown to be positively correlated with synaptophysin immunoreactivity (Sabbagh et al 1998). [3H]Epibatidine binding at autopsy in the temporal cortex has also been demonstrated to be inversely correlated with b-amyloid (Ab) 1– 42 content in the temporal cortex, although not with amyloid plaque or neurofibrillary tangle density in a group of 81 patients with a range of Clinical Dementia Rating from 0 to 5 (Perry et al 2000). This suggests that it is the accumulation of what is generally believed to be the toxic amyloid component contributing to senile plaque formation that is associated with the loss of nicotine binding. In contrast, a positive correlation between [125I]aBGT binding and plaque density was observed in both AD cases and elderly control subjects in the entorhi-nal cortex (Perry et al 2000). The significance of this finding is at present uncertain, in particular in light of the findings suggesting that Ab1– 42 binds with high affinity toaBGT binding sites (Wang et al 2000).
A possible functional link between nAChRs and AD-type pathology is also indicated by two studies comparing cortical amyloid plaque and neurofibrillary tangle densi-ties in tobacco smokers and nonsmokers. In both normal elderly individuals (Perry et al 2000) and groups of unselected cases coming to autopsy (Ulrich et al 1997), plaques but not tangles were significantly fewer in tobac-co-smoking groups. A number of case-control and fol-low-up studies have also indicated that tobacco use may be associated with a reduced risk of developing AD and a delayed onset of familial AD (for a review, see Lee 1994). However, a more recent population-based study (Rotter-dam) demonstrated a significantly increased risk of devel-oping AD in smokers (Ott et al 1998). There is likely to be a complex relationship between tobacco use and the incidence of dementia, which may be complicated by established adverse effects of smoking on cardio- and cerebrovascular systems and interactions between amyloid
and microvascular pathology in AD (Kalaria 1997). Inves-tigations of the effects of long-term exposure to nicotinic drugs in appropriate animal models are required to con-firm that the differences reported above were not the result of confounding variables. However, the possibility that nAChRs have a role in protecting aging neurons is strongly suggested by the demonstration that transgenic b2 knockout mice develop accelerated age-related brain pathology, in terms of hippocampal neuron loss and enhanced gliosis (Zoli et al 1999).
Conclusions
Although deficits in nAChRs in AD have long been recognized, the extent, subunit specificity, and conse-quences of these phenomena are only now being evalu-ated. The most consistently observed change in nAChR expression in AD is the loss in cerebral cortical areas of thea4 subunit anda4-containing receptors. In addition to the need to examine further subunit deficits in more brain areas in AD to elucidate the potential for targeting nico-tinic therapy, investigation of these deficits should be related to prospectively assessed behavioral and cognitive symptoms in patients. It is likely that some of the variability observed between different studies of relatively small groups of patients—for example, those fora3 and a7 expression—is a reflection of unrecognized differences between subgroups of patients, possibly in terms of different genetic subgroups and noncognitive symptoms.
This work was supported by the Medical Research Council, London. CM-R was a European Community TMR research fellow.
Aspects of this work were presented at the symposium “Nicotine Mechanisms in Alzheimer’s Disease,” March 16 –18, 2000, Fajardo, Puerto Rico. The conference was sponsored by the Society of Biological Psychiatry through an unrestricted educational grant provided by Janssen Pharmaceutica L.P.
References
Alkondon M, Pereira EF, Einsenberg HM, Albuquerque EX (1999): Choline and selective antagonists identify two sub-types of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci 19:2693–2705.
Aubert I, Araujo DM, Cecyre D, Robitaille Y, Gauthier S, Quirion R (1992): Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J Neurochem 58:529 –541.
Aubert I, Poirier J, Gauthier S, Quirion R (1994): Multiple cholinergic markers are unexpectedly not altered in the rat dentate gyrus following entorhinal cortex lesions. J Neurosci 14:2476 –2484.
Alzhei-mer’s disease. In: Katzman R, editor. Biological Aspects of Alzheimer’s Disease. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory, 219 –232.
Breese CR, Adams C, Logel J, Drebing C, Rollins Y, Barnhart Y, et al (1997a): Comparison of regional expression of nicotinic acetylcholine receptor alpha 7 mRNA and [125I]-alpha-bungarotoxin binding in postmortem brain. J Comp Neurol 387:385–398.
Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, et al (1997b): Effect of smoking history on [3
H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther 282:7–13.
Burghaus L, Schutz U, Krempel U, de Vos RA, Jansen Steus EN, Wevers A, et al (2000): Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer’s patients. Brain Res Mol Brain Rev 76:385–388. Chen S, Bentivoglio M (1993): Nerve growth factor receptor-containing cholinergic neurons of the basal forebrain project to the thalamic reticular nucleus of the rat. Brain Res 606:207–221.
Clarke P (1995): Nicotinic receptors and cholinergic neurotrans-mission in the central nervous system. Ann N Y Acad Sci 757:73– 83.
Clarke PB (1992): The fall and rise of neuronal alpha-bungaro-toxin binding proteins. Trends Pharmacol Sci 13:407– 413. Clarke PB (1993): Nicotinic receptors in mammalian brain:
Localization and relation to cholinergic innervation. Prog Brain Res 98:77– 83.
Clarke PB, Reuben M (1996): Release of [3H]-noradrenaline from rat hippocampal synaptosomes by nicotine: Mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. Br J Pharmacol 117:595– 606.
Clarke PBS, Pert A (1985): Autoradiographic evidence for nicotine receptors on nigralstriatal and mesolimbic dopami-nergic neurons. Brain Res 348:355–358.
Court J, Piggott M, Lloyd S, Cookson N, Ballard C, McKeith I, et al (2000): Nicotine binding in human striatum: Elevation in schizophrenia and reductions in dementia with Lewy bodies, Parkinson’s disease and Alzheimer’s disease and in relation to neuroleptic medication. Neuroscience 98:79 – 87. Court J, Spurden D, Lloyd S, McKeith I, Ballard C, Cairns N, et
al (1999): Neuronal nicotinic receptors in dementia with Lewy bodies and schizophrenia:a-Bungarotoxin and nicotine binding in the thalamus. J Neurochem 73:1590 –1597. Court JA, Lloyd S, Johnson M, Griffiths M, Birdsall NJM,
Piggott MA, et al (1997): Nicotinic and muscarinic cholin-ergic receptor binding in the human hippocampal formation during development and aging. Dev Brain Res 101:93–105. Court JA, Perry EK (1995): Distribution of nicotinic receptors in
the CNS. In: Stone TW, editor. CNS Neurotransmitters and Neuromodulators. London: CRC Press, 85–104.
Cummings JL, Benson DF (1987): The role of the nucleus basalis of Meynert and dementia: Review and reconsideration. Alz-heimer Dis Assoc Disord 1:128 –145.
Davies P, Feisullin S (1981): Postmortem stability ofa -bunga-rotoxin binding sites in mouse and human brain. Brain Res 216:449 – 454.
Flores CM, Da´vila-Garcı´a M, Ulrich YM, Kellar KJ (1997): Differential regulation of neuronal receptor binding sites
following chronic nicotine administration. J Neurochem 69: 2216 –2219.
Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ (1992): A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol 41:31–37.
Flynn DD, Ferrari-DiLeo G, Levey AI, Mash DC (1995): Differential alterations in muscarinic receptor subtypes in Alzheimer’s disease: Implications for cholinergic based ther-apies. Life Sci 56:869 – 876.
Gearing M, Levey AI, Mirra SS (1997): Diffuse plaques in the striatum in Alzheimer’s disease (AD): Relationship to the striatal mosaic and selected neuropeptide markers. J Neuro-pathol Exp Neurol 56:1363–1370.
Guan Z-Z, Zhang X, Ravid R, Nordberg A (2000): Decreased protein levels of nicotinic receptor subunits in the hippocam-pus and temporal cortex of patients with Alzheimer’s disease. J Neurochem 74:237–243.
Hellstrom-Lindahl E, Court JA (in press): Nicotinic acetylcho-line receptors during pre-natal development and brain pathol-ogy in human aging. Behav Brain Res.
Hellstrom-Lindahl E, Mousavi M, Zhang X, Ravid R, Nordberg A (1999): Regional distribution of nicotinic receptor subunit mRNAs in human brain: Comparison between Alzheimer and normal brain. Mol Brain Res 66:94 –103.
Hossmann K (1999): The hypoxic brain. Insights from ischemic research. Adv Exp Biol Med 474:155–169.
Kalaria RN (1997): Cerebrovascular degeneration is related to amyloid-beta protein deposition in Alzheimer’s disease. Ann N Y Acad Sci 826:263–271.
Kellar KJ, Wonnacott S (1990): Nicotinic cholinergic receptors in Alzheimer’s disease. In: Wonnacott S, Russell MAH, Stolerman IP, editors. Nicotine Psychopharmacology: Molec-ular, Cellular and Behavioural Aspects. Oxford, UK: Oxord University Press, 341–373.
Lee PN (1994): Smoking and Alzheimer’s disease: A review of the epidemiological evidence. Neuroepidemiology 13:131– 144.
Le Novere N, Zoli M, Changeux J-P (1996): Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci 8:2428 –2439.
Lindstrom J, Anand R, Peng X, Gerzanich V, Wang F, Li Y (1995): Neuronal nicotinic receptor subtypes. Ann N Y Acad Sci 757:100 –116.
Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgm-eyer I, Heinemann SF, Collins AC (1992): Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci 12:2765–2784.
Martin-Ruiz CM, Court JA, Molnar E, Lee M, Gotti C, Ma-malaki A, et al (1999): a4 but not a3 and a7 nicotinic acetylcholine receptor subunits are lost from the temporal cortex in Alzheimer’s disease. J Neurochem 73:1635–1640. McGehee DS, Heath MJS, Gelber S, Devay P, Role LW (1995):
Nicotine enhancement of fast excitatory synaptic transmis-sion in CNS by presynaptic receptors. Science 269:1692– 1696.
nicotinic acetylcholine receptors expressed by vertebrate neurons. Ann Rev Physiol 57:521–546.
Minoshima S, Cross D, Foster N, Henry T, Kuhl D (1999): Discordance between tradtional pathology and energy meta-bolic changes in very early Alzheimer’s disease. Pathophys-iological implications. Ann N Y Acad Sci 893:350 –352. Miyai I, Ueno S, Yorifuji S, Fujimura H, Tarui S (1990):
Alterations in neocortical expression of nicotinic acetylcho-line receptor mRNAs following unilateral lesions of the rat nucleus basalis magnocellularis. J Neural Transm 82:79 –91. Nordberg A (1992): Neuroreceptor changes in Alzheimer
dis-ease. Cerebrovasc Brain Metab Rev 4:303–328.
Nordberg A (1993): Neuronal nicotinic receptors and their implications in ageing and neurodegenerative disorders in mammals. J Reprod Fertil Suppl 46:145–154.
Nordberg A (1999): PET studies and cholinergic therapy in Alzheimer’s disease. Rev Neurol (Paris) 155(suppl 4):S53– S63.
Nordberg A, Hartvig P, Lilja A, Viitanen M, Winblad B (1990): Decreased uptake and binding of 11C-nicotine in brain of Alzheimer patients as visualized by positron emission tomog-raphy. J Neural Transm Park Dis Dement Sect 2:215–224. Nordberg A, Lundqvist H, Hartvig P, Lilja A, Langstrom B
(1995): Kinetic analysis of regional (S)(2)11C-nicotine bind-ing in normal and Alzheimer brains—in vivo assessment using positron emission tomography. Alzheimer Dis Assoc Disord 9:21–27.
Ott A, Slooter AJC, Hofman A, van Harskamp F, Witteman JCM, van Broeckhoven C, van Duijin CM, Breteler MMB (1998): Smoking and risk of dementia and Alzheimer’s disease in a population-based cohort study: The Rotterdam study. Lancet 351:1840 –1843.
Perry E, Court J, Goodchild R, Griffiths M, Jaros E, Johnson M, et al (1998): Clinical neurochemistry: Developments in de-mentia research based on brain bank material. J Neural Transm 105:915–933.
Perry E, Martin-Ruiz C, Lee M, Griffiths M, Johnson M, Piggott M, et al (2000): Nicotinic receptor subtypes in human brain ageing, Alzheimer and Lewy body diseases. Eur J Pharmacol 393:215–222.
Perry EK, Court JA, Johnson M, Piggott MA, Perry RH (1992): Autoradiographic distribution of [3
H]nicotine binding in hu-man cortex: Relative abundance in subicular complex. J Chem Neuroanat 5:399 – 405.
Perry EK, Court JA, Piggott MA, Perry RH (1994): Cholinergic component of dementia and aging. In: Huppert FA, Brayne C, O’Connor DW, editors. Dementia and Normal Aging. New York: Cambridge University Press, 437– 469.
Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, et al (1995): Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: Possible index of early neuropathology. Neuro-science 64:385–395.
Perry EK, Smith CJ, Perry RH, Johnson M, Fairbairn AF (1989): Nicotine (3H-nicotine) receptor binding in human brain: Characterization and involvement in cholinergic neuropathol-ogy. Neurosci Res Commun 5:117–124.
Piggott MA, Perry EK, Marshall EF, McKeith IG, Johnson M, Melrose HL, et al (1998): Nigrostriatal dopaminergic
activi-ties in dementia with Lewy bodies in relation to neuroleptic sensitivity: Comparisons with Parkinson’s disease. Biol Psy-chiatry 44:765–774.
Prusky GT, Shaw C, Cynader MS (1987): Nicotine receptors are located on lateral geniculate nucleus terminals in cat visual cortex. Brain Res 412:131–138.
Rapoport S (1999): In vivo PET imaging and postmortem studies suggest potentially reversible and irreversible stages of brain metabolic failure in Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci 249:46 –55.
Rinne J, Myllykyla¨ T, Lo¨nnberg P, Marjama¨ki P (1991): A postmortem study of brain nicotinic receptors in Parkinson-son’s disease and Alzheimer’s disease. Brain Res 547:167– 170.
Rubboli F, Court JA, Sala C, Morris C, Chini B, Perry E, Clementi F (1994): Distribution of nicotinic receptors in the human hippocampus and thalamus. Eur J Neurosci 6:1596 – 1604.
Sabbagh M, Reid R, Corey-Bloom J, Rao T, Hansen L, Alford M, et al (1998): Correlations of nicotinic binding with neurochemical markers in Alzheimer’s disease. J Neural Transm 105:709 –717.
Sahin M, Bowen WD, Donoghue JP (1992): Location of nico-tinic and muscarinic cholinergic and mu-opiate receptors in rat cerebral neocortex: Evidence from thalamic and cortical lesions. Brain Res 579:135–147.
Shimohama S, Tanniguchi T, Fujiwara M, Kameyama M (1986): Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer’s-type dementia. J Neurochem 46:288 –293. Spurden D, Court J, Lloyd S, Oakley A, Perry R, Pearson C, et
al (1997): Nicotinic receptor distribution in the human thal-amus: Autoradiographical localization of [3
H]nicotine and [125
I]abungarotoxin binding. J Chem Anat 13:105–113. Sugaya K, Downen M, Giacobini E (1991): Nucleus basalis
lesions decreasea- andk-bungarotoxins binding in rat cortex. Neuroreport 2:177–180.
Sugaya K, Giacobini E, Chiappinelli VA (1990): Nicotinic acetylcholine receptor subtypes in human frontal cortex: Changes in Alzheimer’s disease. J Neurosci Res 27:349 –359. Terzano S, Court J, Fornasari D, Griffiths M, Spurden D, Lloyd S, et al (1998): Expression of the a3 nicotinic receptor subunit mRNA in human brain in aging and Alzheimer’s disease. Mol Brain Res 63:72–78.
Tilson H, Schwartz R, Ali S, McLamb R (1989): Colchicine administered into the area of the nucleus basalis decreases cortical nicotinic cholinergic receptors labelled by [3H]nico-tine. Neuropharmacology 28:855– 861.
Tohgi H, Utsugisawa K, Yoshimura M, Nagane Y, Mihara M (1998): Age-related changes in nicotinic acetylcholine recep-tor subunits alpha4 and beta2 messenger RNA expression in postmortem human frontal cortex and hippocampus. Neurosci Lett 245:139 –142.
Ulrich J, Johannson-Locher G, Seiler WO, Stahelin HB (1997): Does smoking protect from Alzheimer’s disease? Alzheimer-type changes in 301 unselected brains from patients with known smoking history. Acta Neuropathol 94:450 – 454. Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J,
the central nervous system: A hybridization histochemical study in the rat. J Comp Neurol 284:314 –335.
Wang H, Lee D, D’Andrea M, Peterson P, Shank R, Reitz A (2000): Beta-amyloid (1-42) binds to alpha nicotinic acetyl-choline receptor with high affinity: Implications for Alzhei-mer’s disease pathology. J Biol Chem 275:5626 –5632. Warpman U, Nordberg A (1995): Epibatidine and ABT 418
reveal selective losses of alpha 4 beta 2 nicotinic receptors in Alzheimer brains. Neuroreport 6:2419 –2423.
Wenk GL, Rokaeus A (1988): Basal forebrain lesions differen-tially alter galanin levels and acetylcholinergic receptors in the hippocampus and neocortex. Brain Res 460:17–21. Wevers A, Jeske A, Lobron C, Birtsch C, Heinemann S,
Maelicke A, et al (1994): Cellular distribution of nicotinic acetylcholine receptor subunit mRNAs in the human cerebral cortex as revealed by non-isotopic in situ hybridization. Brain Res Mol Brain Res 25:122–128.
Wevers A, Monteggia L, Nowacki S, Bloch W, Schu¨tz U, Lindstrom J, et al (1999): Expression of nicotinic acetylcho-line receptor subunits in the cerebral cortex in Alzheimer’s
disease-histotopographical correlation with amyloid plaques and hyperphosphorylated tau-protein. Eur J Neurosci 11: 2551–2565.
Wilkie GI, Hutson P, Sullivan JP, Wonnacott S (1996): Pharma-cological characterization of a nicotinic autoreceptor in rat hippocampal synaptosomes. Neurochem Res 21:1141–1148.
Wonnacott S (1997): Presynaptic nicotinic ACh receptors. Trends Neurosci 20:92–98.
Xuereb JH, Perry EK, Candy JM, Bonham JR, Perry RH, Marshall E (1990): Parameters of cholinergic neurotransmis-sion in the thalamus in Parkinson’s disease and Alzheimer’s disease. J Neurol Sci 99:185–197.
Zoli M, Lena C, Picciotto M, Changeux J-P (1998): Identifica-tion of four classes of brain nicotinic receptors using beta2 mutant mice. J Neurosci 18:4461– 4472.
![Figure 1. [3hippocampal formationmusgeniculate nucleus. Autoradiography wasaccording to Court et al (1997) and Spurdenet al (1997) using 1.2 nmol/L [bungarotoxin bindingH]Nicotine (A, C) and [125I]�- (B, D) in the human (A, B) and thala- (C, D) at the leve](https://thumb-ap.123doks.com/thumbv2/123dok/3142593.1383349/2.612.62.380.79.338/hippocampal-formationmusgeniculate-autoradiography-wasaccording-spurdenet-bungarotoxin-bindingh-nicotine.webp)
![Table 3. [125I]�-Bungarotoxin Binding in the Hippocampus](https://thumb-ap.123doks.com/thumbv2/123dok/3142593.1383349/4.612.123.488.521.683/table-i-bungarotoxin-binding-in-the-hippocampus.webp)
![Table 5. [3H]Nicotine Binding in the Thalamus](https://thumb-ap.123doks.com/thumbv2/123dok/3142593.1383349/5.612.62.295.497.660/table-h-nicotine-binding-in-the-thalamus.webp)
![Figure 2. [3H]Nicotine binding and dopamine levels in thecaudate nucleus in Alzheimer’s disease (AD) and dementia withLewy bodies (DLB), derived from Court et al (in press).Subgroups of patients taking neuroleptic medications and thosefree of neuroleptics](https://thumb-ap.123doks.com/thumbv2/123dok/3142593.1383349/6.612.121.487.499.651/nicotine-thecaudate-alzheimer-subgroups-neuroleptic-medications-thosefree-neuroleptics.webp)
