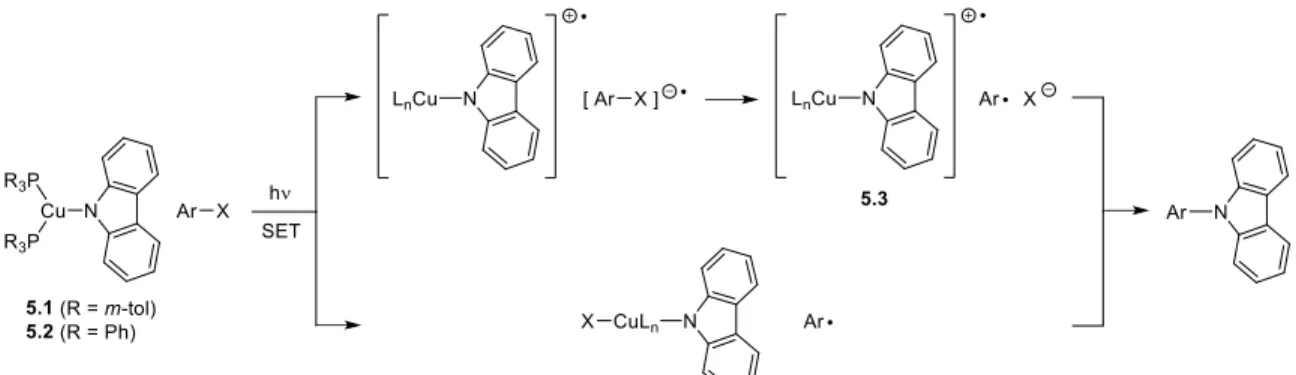
I have had the opportunity to explore many of the off-trail pieces of California and the Southwest in general, and I am extremely grateful to those who have accompanied me on these explorations. The mechanism likely proceeds via single-electron transfer from an excited state of the copper complex to the aryl halide, generating an aryl radical.
Introduction
Motivation
Earth-abundant transition metals in catalysis
- Copper coupling catalysis and photochemistry
The complementarity of thermochemical and photochemical conditions is well recognized for a variety of catalyzed and uncatalyzed reactions.10-12 The application of photolysis to catalytic reactions, often in conjunction with a metal catalyst, has proven extremely fruitful in achieving otherwise unattainable reactivity. 13 Often, first-row metal catalysts are particularly suitable for these reactions because of their propensity to undergo the one-electron type of reactions that are also a hallmark of photoredox chemistry.14,15 One of projects discussed in this thesis (Chapter 5) exploit this synergy to develop photolytic, Cu-mediated and Cu-catalyzed C-N coupling reactions. Thus, this work demonstrates the use of photochemical reaction conditions to help direct the reactivity of first-row transition metals toward desirable processes.
Controlling metal reactivity through ligand design
- Bioinspired ligand and complex design
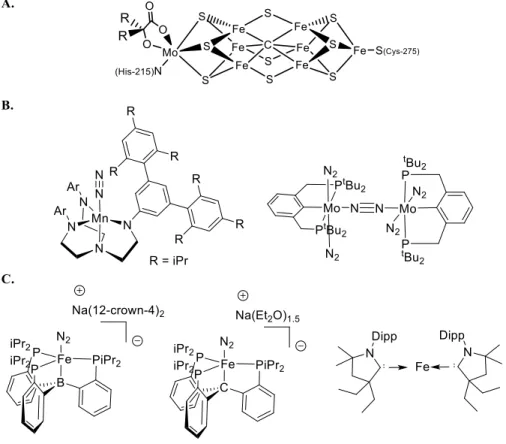
Metalloenzymes exhibit an incredible ability to tune the reactivity of earth-abundant metals through exquisite design of the primary and secondary coordination spheres to perform challenging small-molecule activation reactions; this aptitude has yet to be fully matched by synthetic chemists. Incorporating such interactions into synthetic systems has proven very fruitful, especially in the context of small molecule activation, as it may be.
![Figure 1.2. Biomimetic and bioinspired synthetic metal complexes. (A) Synthetic model of the [FeFe] hydrogenase active site](https://thumb-ap.123doks.com/thumbv2/123dok/10406763.0/29.918.154.812.326.439/figure-biomimetic-bioinspired-synthetic-complexes-synthetic-hydrogenase-active.webp)
Nitrogen activation by molecular systems
- The Mechanism of N 2 Fixation by Molecular Iron Systems
- Analogy to nitrogenase
General schematic of the limiting pathways (distal and alternating) proposed for the conversion of N2 to 2NH3 at a single metal center. Taken together, these facts provide strong support that A is a true intermediate in the catalytic cycle, and suggest that at least the beginning of the cycle follows a distal, Chatt-type pathway (Figure 1.5).
Chapter summaries
In this sense, one of the principles of bioinorganic modeling chemistry is that there can be a feedback mechanism between enzymes such as nitrogenase, various hydrogenases, photosystem II, etc., and small inorganic molecules that act as structural and/or functional models, whereby model chemistry can provide insight into the mechanism of a biological system, and consequently a better understanding of the structural elements of metalloenzymes can be incorporated into synthetic systems to obtain more efficient catalysts. Some of the research presented in this thesis is directed in this direction, as described below.
The ligands used—tridentate tris(phosphine)borate ligands, which enforce a pseudotetrahedral geometry, and phosphiniminato ligands, which exhibit remarkable electronic flexibility—act together to enable a four-coordinate Fe(II) center to display unusual spin crossover properties.
Introduction
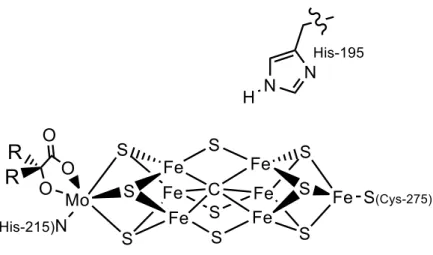
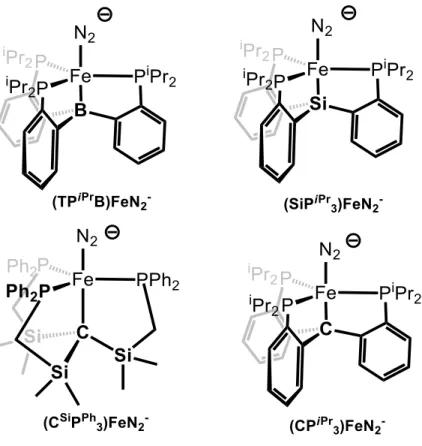
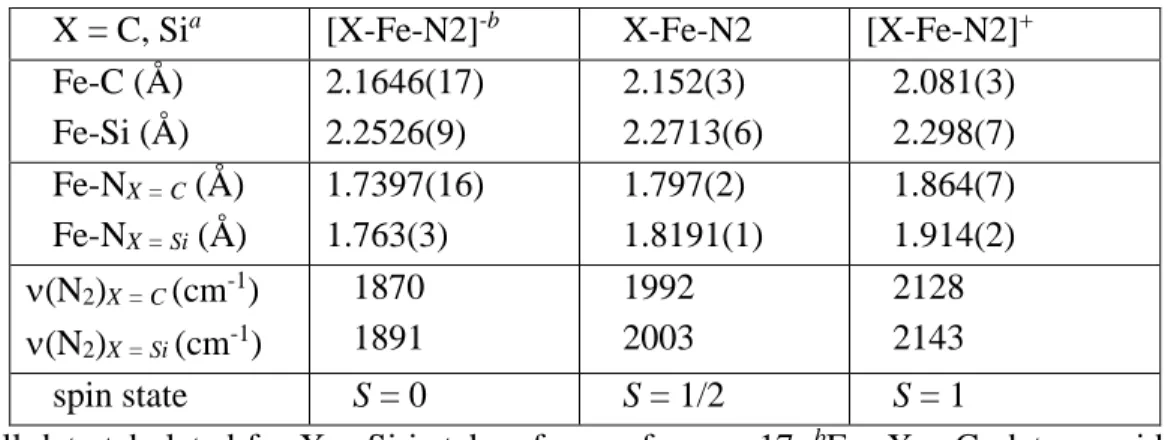
In designing these scaffolds, we have hypothesized that the proposed flexibility of the Fe-C bond in the FeMo cofactor may be facilitated by the ability of the environment around the interstitial carbide—five additional electropositive Fe atoms—to stabilize the development of negative charge on carbon. To this end, we set about the synthesis of the new ligand (CPiPr3)H and the development of its Fe-N2.
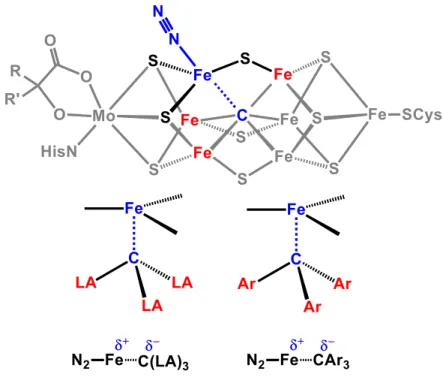
Results and Discussion .1 Ligand Synthesis .1 Ligand Synthesis
- Metallation at iron and precursor complexes
- Reactivity studies
- Physical methods
- X-ray Crystallography
- Computations
- Syntheses
- Reactions
The solid was washed copiously with acetonitrile and then dried in vacuo to yield 1.4 g (2.36 mmol, 68%) of the desired product. Satisfactory elemental analysis could not be obtained due to the lability of the coordinated N2 ligand. After completion of the vacuum transfer, the flask was capped and warmed to room temperature.
Any remaining acid was dissolved in cold Et2O (0.25 mL) and then added, and the Schlenk tube was capped. The reaction was allowed to stir for 60 min at −78 °C before being warmed to room temperature and stirred for 15 min. Thiolate-bridged diiron complexes that bind N2 in the FeIIFeII, FeIIFeI, and FeIFeI redox states and the FeIFeI redox states and the FeIFeI redox states.
Introduction
Results and Discussion
Synthesis of protected ligand 3.4
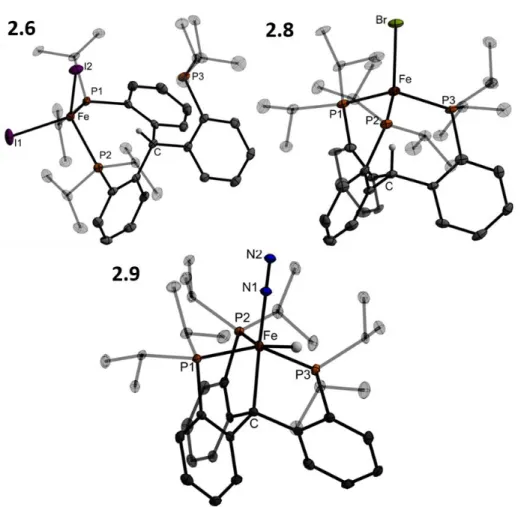
Deprotection of the isopropyl thioether with sodium naphthalenide affords the thiolate ligand 3.5 which, when mixed with two equivalents of FeCl2, generates a metallic brown paramagnetic solid formulated as 3.6 (Scheme 3.2). The two SiP2FeCl subunits are bent with respect to the central ring of the arene; the Cl-Fe-Fe-Cl dihedral angle is 36o. Both salts of the formal diiron(I) 3.8 anion are deep green in color and diamagnetic due to strong coupling.
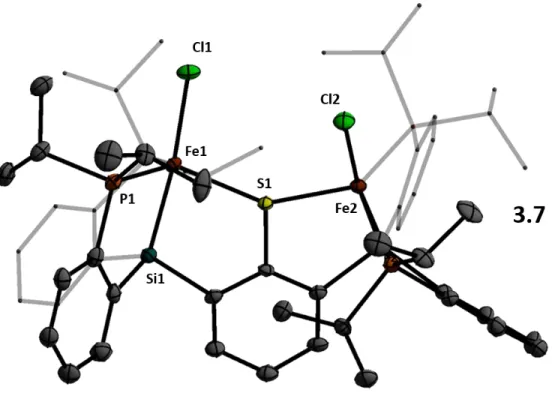
Synthetic access to hydrides 3.11 and 3.12
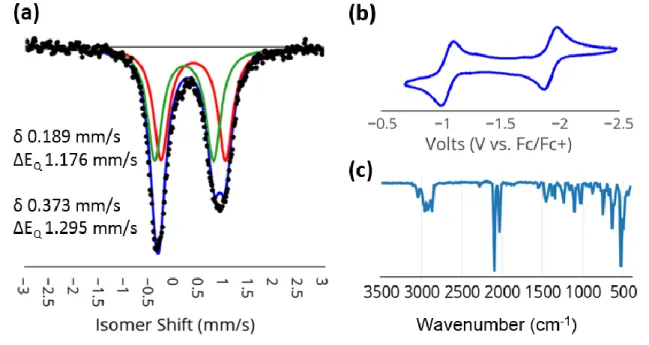
-Vis data unequivocally confirm the different identities of crystals of the two species; 3.11 and 3.9 are also not readily interconverted by any known reaction or decomposition. a) Mossbauer spectrum of microcrystalline 3.11 (80 K, suspended in boron nitride matrix). To chemically confirm the presence of the hydride ligand, 3.11 was exposed to an atmosphere of CO2 in benzene. This variation reflects the presence of a hydride ligand at Fe1, apparently in a position trans to the bridged thiolate donor as assigned to the solution structure of diamagnetic 3.11.
Concluding Remarks
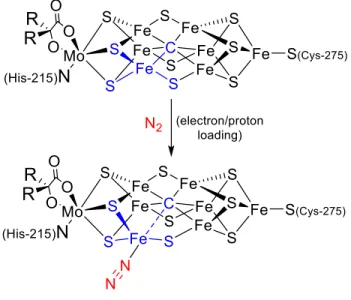
Furthermore, this subunit shows that the N2 ligands are retained in at least three redox states (FeIIFeII, FeIIFeI, FeIFeI) in the presence of the thiolate donor. This is significant because formally low-grade iron sites in the presence of S2 or SH are plausible intermediates of biological nitrogen fixation, but are not well represented in the synthetic literature. Synthetic access to terminally bound iron hydrides in the presence of the bridging thiolate and N2 ligands has also been established.21 Finally, the ability of the present scaffold to mediate the stoichiometric and catalytic cleavage of N-N bonds has been briefly investigated.
Experimental Procedures .1 General considerations
- Physical methods
- Syntheses and characterization data
Fourier transform infrared ATR spectra were collected on a Thermo Scientific Nicolet iS5 spectrometer with diamond ATR crystal (iD5 ATR insert used). Solution-phase magnetic measurements were performed by the method of Evans. 24 Mossbauer spectra were recorded on a SEE Co. spectrometer. Edina, MN) that operated in constant acceleration mode in a transmission geometry.
Synthesis of 3.1 from 2,6-dibromoaniline
Reactivity studies
The solid residue was treated with a KOtBuin THF solution and again the vac was transferred to the HCl solution. The resulting ammonium chloride was analyzed by the indophenol method.26 This experiment was repeated twice, yielding 1.5 and 2.1 equivalents of ammonia. After one hour, the volatiles were transferred to ethereal HCl (4 mL, 2 M) and the resulting ammonium chloride.
While modest yields of NH3 were sometimes obtained without added acid, it was highly irreproducible. Bulletin of the Academy of Sciences of the USSR, Department of Chemical Sciences (English translation).
Introduction
In most cases, these model complexes lack the peripheral secondary sphere interactions present in biological systems. Structural, computational and mechanistic studies on metalloenzymes show that the vast majority of known examples probably benefit from the presence of secondary sphere interactions such as hydrogen bonds to facilitate catalytic reactivity at the metal center - both through the stabilization of intermediates and through the necessary arrangement of reactants .2. Inorganic chemists have increasingly worked to incorporate secondary sphere interactions, especially hydrogen bonding, into structural and functional models of metalloenzyme active sites.
Results and Discussion
- Ligand Design and Synthesis
The influence of pendant amine groups on the reactivity of Fe-N2 complexes with protons is also studied.
Synthesis of ligand arm L 0
Synthesis of ligands L 1 and L 2
Synthesis of Fe(II) and Fe(I) precursor complexes
- Synthesis of Precursor Iron Complexes of L 1 and L 2
- Synthesis and characterization of Fe-N x H y complexes
- Synthesis and reactivity of reduced Fe-N 2 complexes
- Synthesis of a boratrane ligand variant
Structural analysis of and 4.4 clearly illustrates the presence of hydrogen bonding interactions between the tertiary amines in the ligand and the N-H bonds of the iron-bound substrates (Figure 4.4). In the case of ammonia complex 4.3', two independent molecules are present in the asymmetric unit. In the case of 4.6', the N-Na bonds are long enough that the six-membered acecycle can maintain a distorted chair conformation while still supporting this interaction.
![Table 4.1. Comparison data for [Fe]Cl and [Fe]N 2 complexes 17b,c](https://thumb-ap.123doks.com/thumbv2/123dok/10406763.0/128.892.168.759.139.609/table-comparison-data-fe-cl-fe-complexes-17b.webp)
Synthesis of tris(phosphine) borane ligands with three (a) or one (b) pendant amines
Conclusions
- Physical methods
- Synthetic Procedures and Characterization Data
- Reactivity studies
This mixture was stirred at room temperature for one hour and then concentrated in vacuo with mild heating (50 oC). This reaction mixture was stirred for one hour and then concentrated to dryness in vacuo. The reaction mixture was concentrated to dryness and the orange residue was extracted with ether and filtered through Celite.
A similarly cooled solution of HBAr4F.2Et2O (63 mg, 0.062 mmol) in 2 mL of Et2O was added in one portion with stirring, resulting in an immediate color change to yellow. The resulting yellow residue was analyzed by NMR, which was consistent with the formation of iron hydride ((SiPiPr2PNMe)Fe(N2)(H)) as the major product. A similarly cooled solution of [HNEtiPr2][BAr4F] (12.8 mg, 0.012 mmol) in 2 mL of Et2O was added in one portion with stirring, resulting in an immediate color change to yellow.
Development of photoinduced, copper-mediated and copper-catalyzed C- N coupling reactions N coupling reactions
Introduction
Despite the enormous importance of copper-based Ullmann C-N coupling reactions, the understanding of the mechanism of these processes has evolved only slowly.6-9,12 It is believed that Ullmann couplings generally initiate with the formation of Cu-N bonds ; however, a variety of pathways have been proposed for the subsequent cleavage of the Ar-X bond, including coordinated oxidative addition and a single electron transfer mechanism (SET, which involves the transfer of halogen atoms) involving radical intermediates (Fig. 5.1 B). In this report we address this shortcoming by providing a series of experimental data for the reaction of aryl halides with a well-defined copper(I) amido complex (5.1), all of which are present. are consistent with a SET/radical mechanism for C-N bond construction. In addition to providing the strongest evidence to date for the viability of a SET pathway for copper-based Ullmann C–N bond formation, this study provides the first examples of a photoinduced variant of this powerful transformation.
Results and discussion
- Stoichiometric C aryl -N coupling reactivity
- Mechanistic probes
- Catalytic C aryl -N coupling reactivity
- Photoinduced, copper-catalyzed C alkyl -N coupling and mechanistic studies Encouraged by the successful coupling of an alkyl electrophile and carbazolide Encouraged by the successful coupling of an alkyl electrophile and carbazolide
Bromobenzene also undergoes Ullmann coupling when irradiated with a CFL of 13 watts in the presence of a copper-carbazolide complex 5.1. When copper-carbazolide complex 5.1 is photolyzed in the presence of a 1:1 mixture of 1-bromoaphthalene and 4-chlorobenzonitrile, Ullmann coupling product 5.13, derived from 4-chlorobenzonitrile, predominates (Fig. 5.8). Finally, when aryl iodide 5.7 (Fig. 5.7) and lithium carbazolide are used as coupling partners in the presence of copper-carbazolide complex 5.1 under the catalytic conditions outlined in Table 5.2, a 1:1 mixture of diastereomers 5.8 and 5.9 is formed (and no uncyclized products ).
Conclusion
In contrast, chemical oxidation of 5.11 with 0.5 equivalents of magic blue without added carbazolide generates a different, broad axial EPR spectrum qualitatively similar to that shown in Figure 5.4, accompanied by a light blue color (Figure 5.14). Continued work since then has expanded the substrate range for photoinduced copper-catalyzed coupling reactions to a wide range of N, O, S, and C nucleophiles.33 Mechanistic studies of these systems also continue, and it is becoming clear that in catalytic systems the mechanism is highly substrate dependent and probably more complex than our original working hypothesis (Figure 5.10). In particular, the transfer of photoelectrons from the nucleophile (e.g. lithium carbazolide) to the electrophilic substrate likely plays an important role under catalytic conditions.
Experimental details
- General
- X-ray crystallography
- Syntheses, characterization data, and reactions
The reaction mixture was then exposed to air, diluted with diethyl ether (15 mL) and washed with distilled water (3X5 mL). The aqueous fractions were re-extracted with 5 mL of diethyl ether and the combined organic layers were washed with 5 mL of saturated aqueous sodium chloride and then dried over magnesium sulfate, filtered and concentrated. The reaction mixture was then diluted with ∼100 mL of diethyl ether, causing white solids to fall out.
Isotopomers formed in synthesis of deuterated radical clock 5.7
In introductory organic chemistry textbooks, the alkylation of amines with alkyl halides is often the first reaction of amines described. There are isolated reports of N-alkylations of amines with unactivated alkyl halides occurring in the presence of a substoichiometric amount of a transition metal. A non-negligible product formation under irradiation in the absence of CuI (16% versus 76% for cyclohexyiodide) may be due to the photophysical properties of lithium carbazolide itself.
Introduction
![Figure 1.5. Mechanistic outline for the catalytic reduction of N 2 to ammonia by [TPBFe]](https://thumb-ap.123doks.com/thumbv2/123dok/10406763.0/35.918.167.796.415.614/figure-mechanistic-outline-catalytic-reduction-n-ammonia-tpbfe.webp)
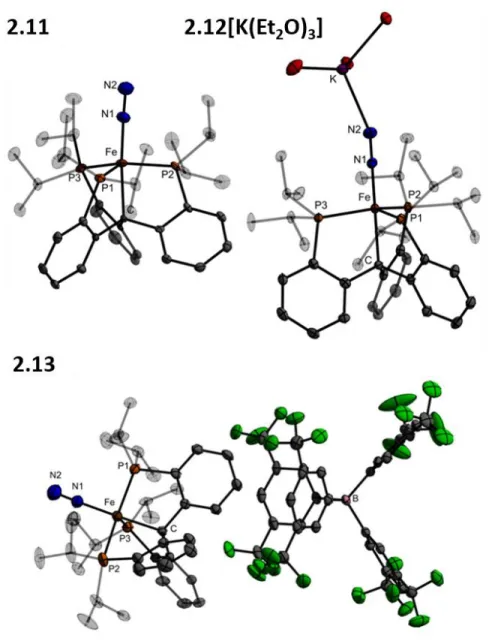
![Figure 2.7. (A) Isocontour plot of the Fe-C alkyl σ bond of 2.12[K(Et 2 O) 3 ] located from NBO analyses](https://thumb-ap.123doks.com/thumbv2/123dok/10406763.0/66.918.257.730.105.435/figure-isocontour-plot-alkyl-bond-located-nbo-analyses.webp)
![Figure 2.8. Spectroscopic analyses of reaction mixtures following the catalytic produc- produc-tion of NH 3 using 2.12[K(Et 2 O) 0.5 ] as a catalyst](https://thumb-ap.123doks.com/thumbv2/123dok/10406763.0/69.918.287.707.112.643/figure-spectroscopic-analyses-reaction-mixtures-following-catalytic-catalyst.webp)
