My time at Caltech was truly enriched by all the wonderful people and scientists I had the opportunity to meet. Brian in particular was a constant source of encouragement to all members of the Reisman lab. The utility of these transformations is highlighted in the five-step syntheses of the natural products (+)-naseaseazine A and B.
LIST OF ABBREVIATIONS
INTRODUCTION
Tryptophan and unnatural tryptophan derivatives are important building blocks in the total synthesis of natural products as well as for the development of new drugs,1 biological probes,2 and chiral small-molecule catalysts.3 The central tryptophan motif can be found in numerous biologically active natural products, either explicitly or implicitly, some of which are shown in Figure 1.1. Because of their promising medicinal relevance, these products have inspired innovative work with new synthetic methods to access the central pyrroloindoline framework, culminating in the total synthesis of a number of these challenging natural products.5 Figure 1.1. The following introductory chapter serves to briefly summarize and highlight modern synthetic strategies and tactics to access unnatural tryptophan derivatives as well as pyrroloindoline alkaloids with selected examples in total synthesis.
SYNTHESIS OF TRYPTOPHAN DERIVATIVES
In 1980, Townsend and co-workers demonstrated that subjecting 6-methyl dehydrotryptophan to [Rh(COD)Cl]2, copper-phosphine complex 10 and 45 psi hydrogen gas gave 6-methyl tryptophan (9) in high enantiomeric excess. (Scheme 1.1, a).7 Subsequent work on asymmetric hydrogenation has further improved this process to provide excellent ee's at low Rh catalyst loadings, making it an efficient choice in many cases. One such enantiospecific approach utilizes orthoiodoanline lines (14) in conjunction with an amino acid-derived coupling partner (Scheme 1.2, a).9 In 1999, Cook and co-workers reported the Pd(0)-catalyzed heteroannulation (Larock indole synthesis ) of o-iodoaniline with Schöllkpf-derived triethylsilyl alkyne auxiliary 15. A complementary approach to Larock's indole synthesis was reported by Jia and Zhu in 2005, using an aldehyde coupling partner (17) instead of a disubstituted alkyne (2 ,b1). 10. Acting via aldimine mediation, Pd-mediated heteroannulation affords unsubstituted 2-tryptophans in moderate to good yields.
TRYPTOPHAN DERIVATIVES IN TOTAL SYNTHESIS
Cook and co-workers also used their methodology in their total synthesis of the complex polycyclic alkaloid alstophylline (Scheme 1.4, b). In 2009, Baran and co-workers reported the total synthesis of capacachin B using Larock's indole synthesis to assemble the key tryptophan motif.17 Starting with tryptophan-derived peptide 24, subjection to o-iodoaniline and NIS provided pyrroloindoline 35 as one diastereomer. Larock's equally impressive indole synthesis was used as a key step in Boger's wonderful synthesis of chloropeptins (Scheme 1.6).
STRATEGIES FOR THE SYNTHESIS OF PYRROLOINDOLINES
As shown in Scheme 1.10, the addition of N-bromosuccinimide and pyridinium p-toluenesulfonate to tryptophan 54 leads to the clean formation of bromopyrroloindoline 58. Using the imidazolinone catalyst 61 with acrolein as an electrophile, a highly enantioselective preparation of C3-alkylated pyrroloindolines was achieved (Scheme 1.11).26. Activation of Michael acceptors can also be performed using chiral Brønsted acids such as (R)-TRIP (Scheme 1.12).
PYRROLOINDOLINES IN TOTAL SYNTHESIS
C–N bond formation followed by Red-Al treatment afforded the natural product psychotrimine (79) in excellent overall yield. A similar approach was taken by Stephenson and co-workers in their synthesis of glyocladin C using photoredox catalysis.30 After oxidative cyclization of protected tryptophan 87, bromopyrroloindoline underwent amidation to form carboximide 89. An additional five steps are then required to construct the diketopiperazine moiety and result in deoxygenation, to provide a natural product.
CONCLUSIONS
INTRODUCTION
Although subsequent reports were able to catalyze this reaction by the addition of stoichiometric phenol derivatives, these complexes have never before been applied to tandem conjugate addition/asymmetric protonation reactions. Despite the prevalence of conjugate addition/asymmetric protonation reactions in the literature, the first report of a Friedel–Crafts conjugate addition/asymmetric protonation reaction was not disclosed until 2008. In particular, there are no examples of Friedel–Crafts conjugate addition/asymmetric protonation reactions. using indole-based nucleophiles to give tryptophan derivs.
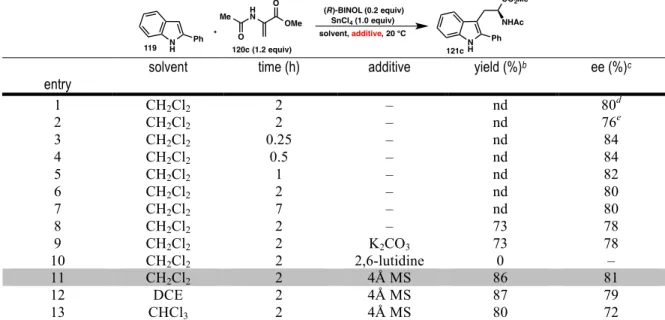
SCREENING AND OPTIMIZATION
As the screening process progressed, we began to notice inconsistencies in the selectivity of the reaction. Instead, the use of activated 4Å molecular sieves increased the yield and selectivity of the reaction, providing tryptophan 121c in 86% yield and 81% ee. Further increasing the steric bulk of the catalyst by substitution with phenyl groups decreased the reactivity and selectivity (entry 6).
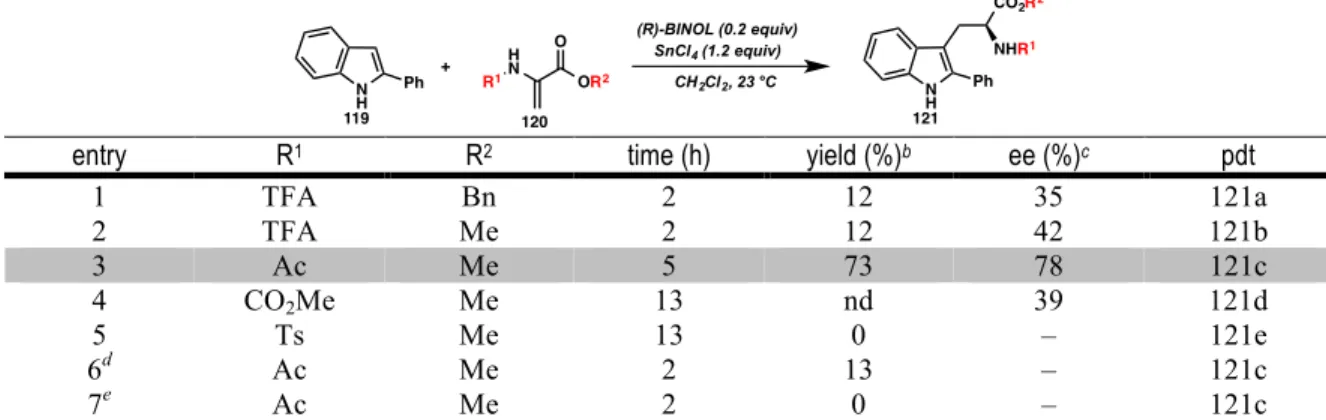
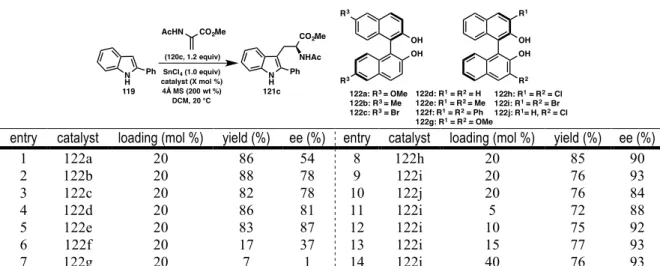
SUBSTRATE SCOPE
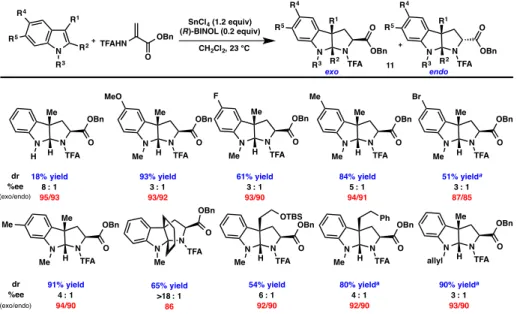
We found that the reaction was amenable to substrates with both alkyl and aryl substitution at the 2-position of the indole. 2-Aryl indoles bearing both electron-donating and electron-withdrawing substituents in the para and meta positions were tolerated (121o-121s). Unfortunately, even small functionality at the ortho position, such as fluorine, resulted in decreased reactivity (121r); a slightly larger methyl group further weakened both yield and ee (121n).
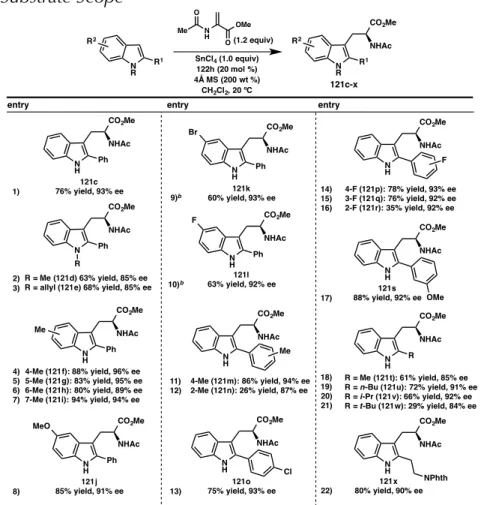
MECHANISTIC STUDIES
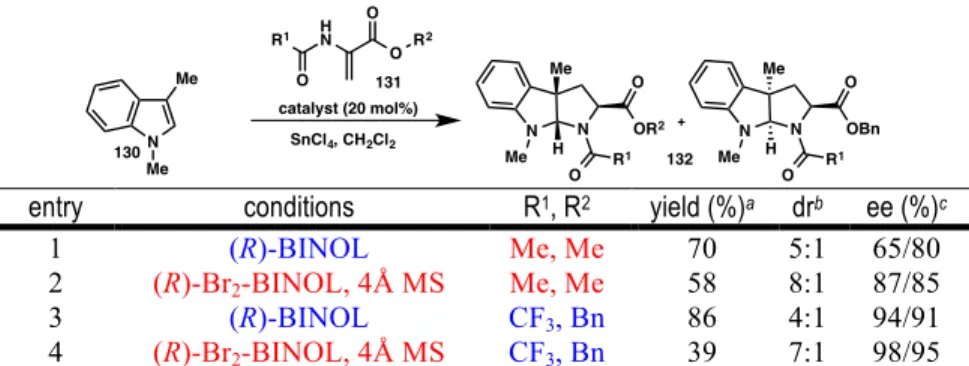
Due to the apparent mechanistic similarities between the formal (3+2) cycloaddition and Friedel-Crafts, we wondered whether our newly optimized conditions for Friedel-Crafts could be applied to the synthesis of pyrroloindolines to increase selectivity. Although there was a noticeable increase in the selectivity of the product mixture compared to the conditions originally reported for acrylate 131 (point 1), better results were still obtained using benzyl 2-trifluoroacetamido acrylate (point 3). Interestingly, the use of acrylate 131 with optimal Friedel–Crafts conditions afforded the product in good dr and excellent enantioselectivity (entry 4).
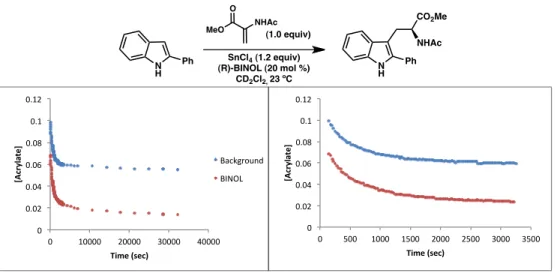
CONCLUSION
EXPERIMENTAL SECTION
The aqueous layer was extracted with EtOAc (45 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated. The combined organics were washed with saturated aqueous NaHCO3 (2 x 45 mL) and brine (45 mL), then dried (Na2SO4), filtered and concentrated. InBr3 (16 mg, 0.05 mmol, 0.05 equiv) was added in one portion and the mixture was heated at 110 ºC for 5 h, then cooled to room temperature, filtered through celite and concentrated.
InBr3 (40 mg, 0.1 mmol, 0.05 equiv) was added in one portion and the mixture was heated to 110 ºC for 5 h, then cooled to room temperature, filtered through celite and concentrated. The aqueous layer was extracted with EtOAc (2 x 5 mL) and the combined organic layers were washed with saturated aqueous NaHCO 3 (5 mL), dried (Na 2 SO 4 ), filtered and concentrated. The aqueous layer was extracted with EtOAc (2 x 5 mL) and the combined organic layers were washed with saturated aqueous NaHCO 3 (5 mL), dried (Na 2 SO 4 ), filtered and concentrated.
The aqueous layer was extracted with EtOAc (2 x 50 mL) and the combined organic layers were washed with saturated aqueous NaHCO 3 (50 mL), dried (Na 2 SO 4 ), filtered and concentrated. The combined organic layers from the acidic aqueous extraction were dried (Na2SO4), filtered and concentrated. The combined organic layers were washed (40 mL H2O, then 40 mL brine), dried (Na2SO4), filtered and concentrated.
The combined organic layers were dried (Na2SO4), filtered and concentrated to give per-deutero-2-phenylindole with 90% deuterium incorporation.
NOTES AND REFERENCES
Sample name: MEK3186-1 Data collected on: indy.caltech.edu-inova500 Archive directory: /home/mkieffer/vnmrsys/data Sample directory: MEK3186-1 FidFile: PROTON01. Sample name: MEK3186-1 Data collected on: indy.caltech.edu-inova500 Archive directory: /home/mkieffer/vnmrsys/data Sample directory: MEK3186-1 FidFile: CARBON01. Sample name: MEK3084-1 Data collected at: indy.caltech.edu-inova500 Archive directory: /home/mkieffer/vnmrsys/data Sample directory: MEK3084-1 FidFile: PROTON01.
Sample name: MEK3084-1 Data collected from: indy.caltech.edu-inova500 Archive directory: /home/mkieffer/vnmrsys/data Sample directory: MEK3084-1 FidFile: CARBON01. Sample name: MEK3193-1 Data collected from: indy.caltech.edu-inova500 Archive directory: /home/mkieffer/vnmrsys/data Sample directory: MEK3193-1 FidFile: PROTON01. Example name: MEK3185-1 Data collected from: indy.caltech.edu-inova500 Archive folder: /home/mkieffer/vnmrsys/data Example folder: MEK3185-1 FidFile: PROTON01.
Sample Name: MEK3150-1 Data Collected at: indy.caltech.edu-inova500 Archive Folder: /home/mkieffer/vnmrsys/data Sample Catalog: MEK3150-1 FidFile: PROTON01. Sample Name: MEK3080-1 Data Collected at: indy.caltech.edu-inova500 Archive Folder: /home/mkieffer/vnmrsys/data Sample Catalog: MEK3080-1 FidFile: PROTON01. Sample Name: MEK3238-1 Data collected at: indy.caltech.edu-inova500 Archive Folder: /home/mkieffer/vnmrsys/data Sample Catalog: MEK3238-1 FidFile: PROTON01.
Sample Name: MEK3113-1 Data Collected at: indy.caltech.edu-inova500 Archive Folder: /home/mkieffer/vnmrsys/data Sample Catalog: MEK3113-1 FidFile: PROTON01. Sample Name: MEK3113-1 Data collected at: indy.caltech.edu-inova500 Archive Folder: /home/mkieffer/vnmrsys/data Sample Catalog: MEK3113-1 FidFile: CARBON01. Sample Name: MEK3079-1 Data collected at: indy.caltech.edu-inova500 Archive Folder: /home/mkieffer/vnmrsys/data Sample Catalog: MEK3079-1 FidFile: PROTON01.
Sample Name: MEK3129-1 Data collected at: indy.caltech.edu-inova500 Archive Folder: /home/mkieffer/vnmrsys/data Sample Catalog: MEK3129-1 FidFile: PROTON01. Sample Name: MEK3129-1 Data collected at: indy.caltech.edu-inova500 Archive Folder: /home/mkieffer/vnmrsys/data Sample Catalog: MEK3129-1 FidFile: CARBON01.
INTRODUCTION
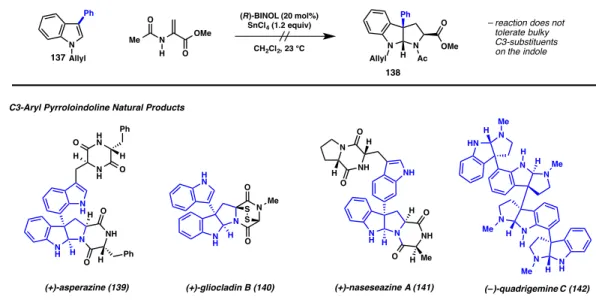
Direct and Selective Copper-Catalyzed Arylation of Tryptamines and Tryptophans: Total Synthesis of (+)-Naseazines A and B†. This finding was particularly unfortunate because of the occurrence of an important subclass of pyrroloindoline natural products characterized by a C3 quaternary center bearing an aryl substituent (Figure 3.1). These compounds, including quadrihemine C (142) and glyocladin B (140), exhibit potent biological activity, but methods for their efficient preparation have remained a challenge in modern synthetic chemistry.4,5 This chapter describes our efforts towards the development of ' a complementary and direct arylation reaction to obtain convergent access to this subclass of natural products.
In a seminal 2001 report, Overman and Govek reported the successful implementation of an intramolecular Heck strategy in the synthesis of (+)-asperazine, a bisindole alkaloid containing a unique C3-C7 aryl bond.6 In 10-step (L)-tryptophan methyl ester hydrochloride was advanced to iodoanilide 143, which in a key step was exposed to Pd2(dba)3, (2-furyl)3P, and PMP to effect a highly diastereoselective, intramolecular Heck reaction to form the C3-arylated quaternary center found in the natural product. The following year, Overman reported the total synthesis of the polypyrroloindoline alkaloid (–)-quadrigemine C, now exploiting a key, enantioselective Heck desymmetrization of a meso compound (Scheme 3.2, b).7 Treatment of meso-146 with Pd(OAc) 2 and ( R)-tol-BINAP with pentamethylpiperidine gives bisoxindole 147, which is efficiently cyclized under reductive conditions to the natural product. A decade later, Movassaghi and co-workers reported a general strategy toward this class of compounds using a bromocyclization/Friedel–Crafts approach (Scheme 1.16, Chapter 1).5c A subsequent publication describes the extension of this strategy toward the completion of indole- with natural products 155–157 (Scheme 3.3).
Again, starting with the tryptophan-derived bromotetracycline 152, subjection to superstoichiometric AgBF4 to generate the tertiary benzylic carbocation followed by addition of four equivalents of an indole nucleophile affords the C3-aryl pyrroloindoline 154. Our strategies presented at the beginning of Overman and Movassaghi represented the state of the art in the preparation of C3-aryl pyrroloindolines. For example, while the Heck reaction is a powerful tool for the generation of quaternary centers, the preparation of the cyclization precursor is lengthy and additional steps are required to advance to pyrroloindolines.
When considering different strategies, we found that there is no direct method for the preparation of C3-arylpyrroloindolines, and therefore we expected that the development of a direct arylation/cyclization cascade of tryptamines and tryptophans would greatly simplify the synthesis and preparation.
REACTION DESIGN
Specifically, the key to the success of this transformation is the generation of C3-metalated species 159, which must undergo reductive elimination and cyclization to provide the desired product (Scheme 3.6). One major concern was the relative stability of such an intermediate, which is known to undergo facile migration to the C2 position of the indole.11 Reductive elimination and rearomatization could then provide 2-aryl indoles. Additionally, one could envision the coordination of a transition metal catalyst with an indole nitrogen or a pendant amine to give Buchwald–Hartwig type products.