The following introductory sections outline our motivation for pursuing the synthesis of cylindrocyclophane natural products via the use of Ni-catalyzed RCC for macrocyclization. These fundamental studies of cyclophane assembly have undoubtedly guided partial and total syntheses 18 of the cylindrocyclphane natural products. The choice of which bond of the aliphatic bridge to construct during macrocyclization can greatly influence the success of cyclization.
![Figure 5.2 [7.7]Paracyclophane natural products](https://thumb-ap.123doks.com/thumbv2/123dok/10400184.0/4.918.234.744.254.569/figure-5-2-7-7-paracyclophane-natural-products.webp)
Cylindrocyclophanes: Stereocenter Installation
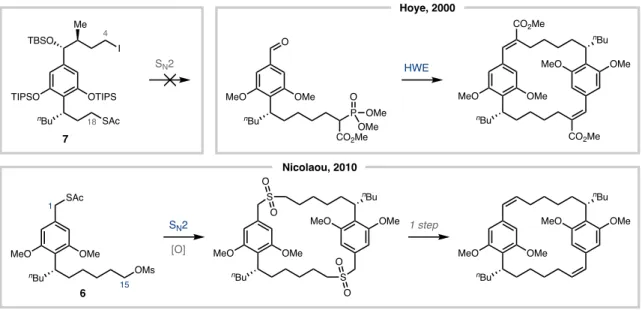
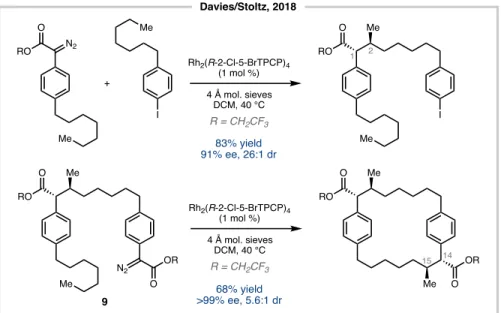
For example, with the syntheses of Hoye68 and Nicolaou69, enantioselective transformations were required to give rise to the trans-C1/C2 hydroxymethyl motif. Hydroboration/oxidation and a sequence of dihydroxylation (and deoxygenation to 2) followed by methylation served this purpose (Figure 5.9). A conceptually different approach was introduced by the Davies/Stoltz report (Figure 5.10).63 A chiral dirhodium catalyst was used for the first time to accommodate the stereodiad C1/C2 with good enantioselectivity and excellent dr.
To develop doubly reductive cross-coupling as a cyclodimerization reaction of 11 , this key event will have to overcome known limitations of single RCCs. In view of preliminary SAR data (vide supra), retrosynthetic cleavage through the C7-nBu bond via simple α-alkylation would be a boon for the preparation of cylindrocyclopane derivatives. We anticipated that our synthetic design would readily lend itself to the preparation of additional cylindrocyclopane variants (eg, 1).

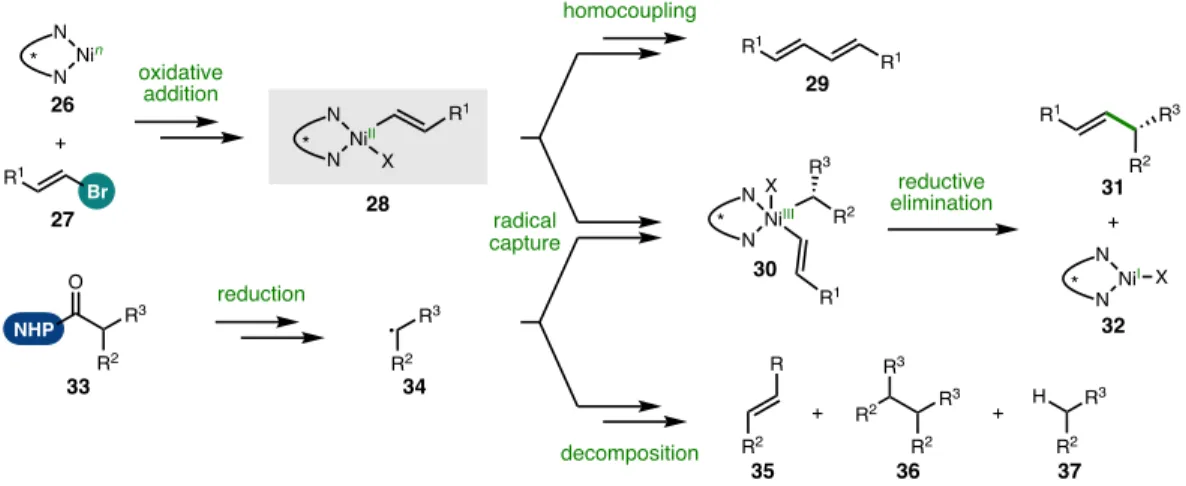
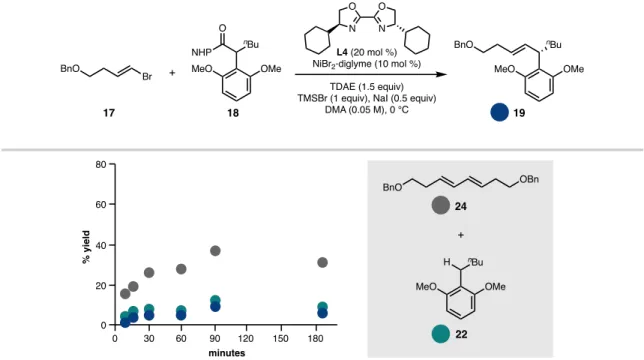
SINGLE REDUCTIVE CROSS-COUPLING: MODEL REACTION Figure 5.12 Motivation for single reductive cross-coupling
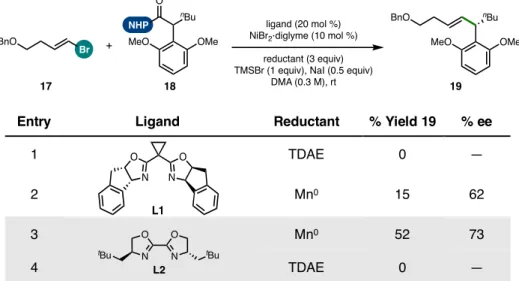
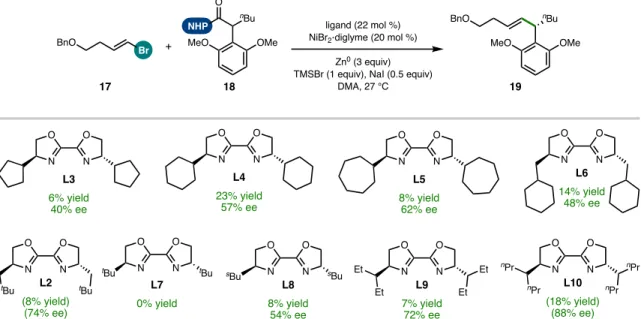
Investigation Using BOX Ligands
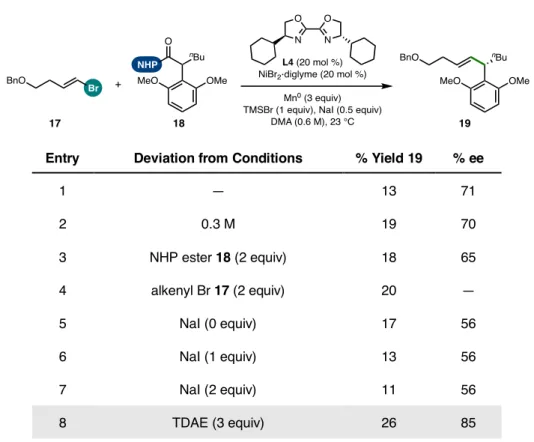
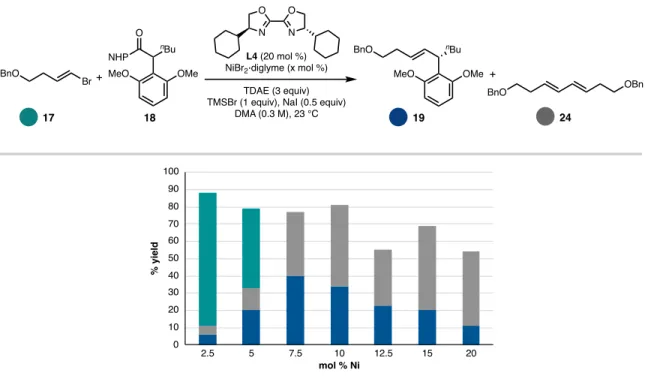
The strong change in the yield of product 19 induced by the BOX ligand framework compared to BiOX (19% yield with L4; 40% yield with L1)—following the optimized order of addition—corresponded with a dramatic decrease in the formation of homocoupled 24 (from 47% This finding suggested that one of the important properties of the IndaBOX (L1) ligand could be the inhibition of alkenyl bromide homocompaction, an effect that could potentially occur through stabilization of the key organonickel intermediate 28 or an oxidative adduct that would be challenging (see Figure 5.15). catalyst loading enabled complete consumption of this substrate with a corresponding improvement in cross-coupled yield (49%).
SYNTHESIS OF DIMERIZATION SUBSTRATES
Toward Cylindrocyclophane F
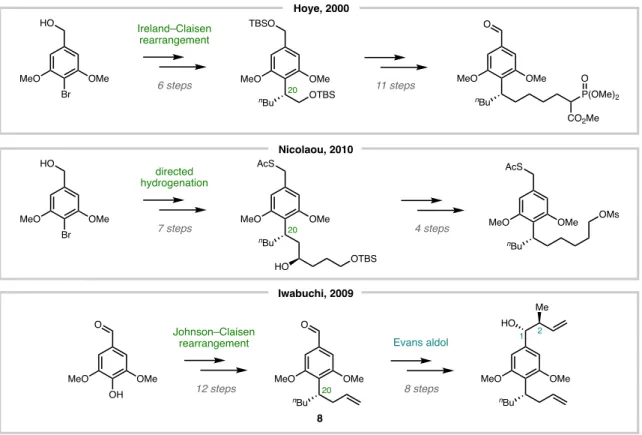
We found that lithiation of the aryl bromide (52) followed by addition of a simple alkyl iodide (54) effected an SN2-type reaction in 71% yield, creating the crucial Csp2–Csp3 bond (Figure 5.26). Instead of the desired coupled product, we observed the protonated arene 50 as the major byproduct, along with Using an alkenyl boronic ester in place of the necessary alkenyl bromide, the putative alkyl radical intermediate (likely generated from alkyl iodide 64) proceeded to side pathways such as 5-exo-trig cyclization to form an α-boryl radical stable, rather than productive linkages (input 2).
Toward Cylindrocyclophane A
In a parallel fashion, we also prepared intermediates designed to investigate the effect of bridge length on [n,n] and [m,m] paracyclophane formation via doubly reductive coupling (Figure 5.37). The cyclophane structures resulting from such unsubstituted monomers would be novel and could be used outside the scope of natural product synthesis (i.e., for host–guest chemistry, as subjects of computational physical organic studies, etc.). Ultimately, however, we planned to return to the system required to access natural product 2 (i.e., methoxy/methyl 83 ) following proof-of-concept macrocyclization.

DOUBLE REDUCTIVE CROSS-COUPLING .1 Guiding Mechanistic Design
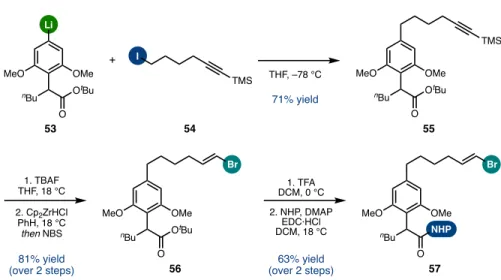
Cyclodimerization versus Polymerization
However, byproducts of the nature observed in the single RCC system were not identified in the crude reaction mixture. A peak in the small molecule regime of the GPC trace matched that of a purified sample of macrocycle 94. Theoretically, the yield of cyclodimer 94 arising from this sequence was limited by the molecular weight and polydispersity of the polymer.
REVISED APPROACH AND FORMAL SYNTHESIS
To move forward towards the synthesis of the cylindrocyclophanes, we considered metathesis depolymerization as a conceptual springboard. In our initial investigation of this custom synthetic design plan, we continued to use the easily accessible, unsubstituted substrate class. This moderate yield is fully consistent with the performance of the truncated NHP ester carrying identical arylo,o dissubstitution in the single RCC model reaction.
Although the entangled dr e 112 remained unknown, we expected that determination of the diastereomeric ratio of the downstream macrocycle could be used as a readout for RCC stereoselectivity. Analysis of the reaction mixture by mass spectrometry allowed us to tentatively assign one of several resulting species as 114. Three observations led us to hypothesize that the aryl substitution pattern of 112, in combination with the position of the internal alkenes (along target bridge.
me. paracyclophane), precludes the efficient reversibility of the various metathesis reactions required to insert into the head-to-tail dimer 113. Consistent with this hypothesis, a possible solution may be to remove the alkenyl methyl substituent and attempt cyclodimerization of di -(terminal) alkene. Desilylation was then accomplished smoothly by treatment of the cross-linked product with TsOH (91% yield), and the enantiomeric excess of the resulting diene (119) was determined to be 96%.
Following the NMR scaling protocol previously established for CM/RCM of the methylated variant (108, see Fig. 5.53), paracyclopane 94 was generated in 49% yield after 30 min of reaction time.
CONCLUDING REMARKS
Direct application of asymmetric reductive cross-coupling conditions to a NHP ester/alkenyl bromide provided the corresponding head-to-tail cyclodimer as a single detectable diastereomer, albeit in less than 10% yield. The propensity for dielectrophilic polymerization under these conditions inspired the development of an unprecedented sequence of reductive polymerization and depolymerization/RCM to deliver the same macrocycle in increased yield with a high level of stereoselectivity. This discovery contributes [7.7]paracyclopanes as novel macrocyclic monomers of reversible polymerization and is an exciting entry into the emerging field of chemical polymer recycling to monomers.150.
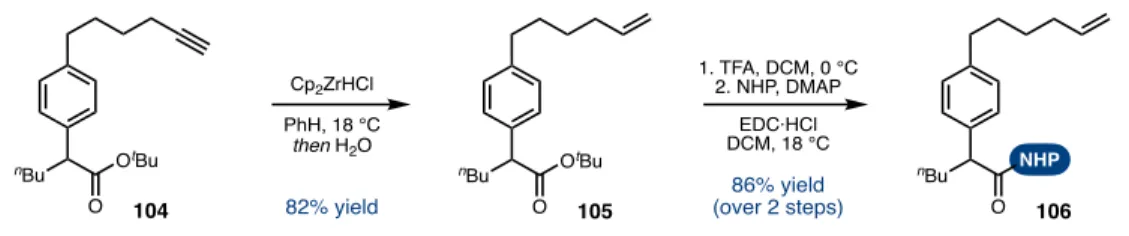
Finally, using Ni-catalyzed reductive Csp3-Csp2 cross-coupling to generate an enantioenriched diene monomer followed by CM/RCM afforded a paracyclopane in good yield. Key CM/RCM precursor 120 was synthesized in ten steps in the longest linear sequence from commercial or known materials and could be progressed to the natural product in two steps.

EXPERIMENTAL SECTION .1 Materials and Methods
Single Reductive Cross-Coupling
The crude residue was purified by column chromatography (silica, 5% EtOAc/hexanes) to give 17 (1.95 g, 85% yield) as a yellow oil. The crude residue was purified by column chromatography (silica, 20% EtOAc/hexanes) to give 18 (1.25 g, 79% yield) as a white amorphous solid. The bottle was capped and then stirred at 1000 rpm outside the glove box at room temperature.
This sample was concentrated and purified by preparative thin layer chromatography (silica, 15% Et2O/hexanes) to afford 19, which was evaluated by chiral SFC. This sample was concentrated and purified by preparative thin layer chromatography (silica, 15% Et2O/hexanes) to provide 19, which was evaluated by chiral SFC. The vial was placed in a temperature-controlled well plate in the glove box and then cooled to −7 °C.
This sample was concentrated and purified by preparative thin layer chromatography (silica, 15% Et 2 O/hexanes) to give 19 which was analyzed by chiral SFC.
Synthesis of Dimerization Substrates tert-butyl 2-(2,6-dimethoxyphenyl)acetate (121)
After complete consumption of the ester, the reaction was quenched by slowly adding water and then the layers were separated. After complete consumption of the ester as judged by TLC, the reaction was allowed to reach room temperature and then concentrated. After complete conversion to acid 123, the reaction was allowed to reach room temperature and diluted with excess sodium.
The reaction was stirred at –78 °C for 30 min, then alkyl iodide 54 (87 mg, 0.31 mmol, 2 equiv) was added via syringe, and the reaction was allowed to warm to ambient temperature and then for 5 h stirred before quenching with sat. The crude residue was purified by column chromatography (silica, 2% Et2O/hexanes) to provide 55 (51 mg, 71% yield) as a colorless oil. The reaction mixture was allowed to stir at 0 °C for 1 h before being quenched with brine and extracted three times with EtOAc.
After complete consumption of the aryl bromide as judged by TLC, the reaction was allowed to reach room temperature and then quenched with water. The crude residue was purified by column chromatography (silica, 5 to 20% EtOAc/hexanes) to afford 80 (134 mg, 93% yield) as a colorless oil. The reaction was stirred at room temperature for 20 h until the solution turned gold when water was added.
The crude residue was purified by column chromatography (silica, 5% EtOAc/hexanes) to afford alkyne 138 (110 mg, 50% yield over 2 steps) as a colorless oil.
Double Reductive Cross-Coupling General Procedure E
Outside the glove box, the mixture was stirred until all solids dissolved and then heated to 50°C in a metal reaction block. Second, instead of silica filtration, the reaction was diluted with PhH (1.0 ml) in the glove box and then transferred via pipette to a 2 dram vial with a stir bar and Teflon cap, and rinsed with PhH (1.5 ml). EtOAc/Hexanes; the filtrate was concentrated and tested by quantitative 1 H NMR to determine that the formation of 94 occurred in 18% yield.
The catalyst stock solution was transferred to the second vial via syringe followed by alkenyl bromide (0.1 mmol, 1 equiv), then the reaction mixture was stirred at ambient temperature at 250 rpm to ensure complete dissolution of the NHP ester and adequate mixing of all reagents (the catalyst is possibly not completely dissolved at this temperature). The crude residue was purified by preparative thin layer chromatography (silica, hexanes) to give 108 (21 mg, 79% yield, 97:3 E/Z) as a colorless oil. The reaction mixture was stirred at ambient temperature until all reagents were dissolved (approximately 1 min) and then transferred via syringe to an NMR tube which was capped and sealed with electrical tape.
At various time points, the reaction was allowed to cool to ambient temperature, evaluated by quantitative 1H NMR to determine the yield of 94, then returned to the hot oil bath. Outside the glove box, the mixture was stirred until all solids were dissolved and then heated to 50 °C in an oil bath. The crude residue was purified by column chromatography (silica, 20% PhMe/hexanes) to give 112 (16 mg, 47% yield) as a colorless oil.
The crude residue was purified by preparative thin layer chromatography (silica, hexanes) to give 144 (27 mg, 83% yield) as a colorless oil.
Development and performance of a Ni(II)-catalyzed reductive cross-coupling of substituted 2-chloropyridine and ethyl 3-chloropropanoate. Nickel-Catalyzed Reductive Cross-Couplings: New Opportunities for Carbon-Carbon Bond Formations by Photochemistry and Electrochemistry. Total synthesis of the spiculisporic acids, progress towards the total synthesis of cylindrocyclopane F, and formal synthesis of cylindrocyclopane A.
Total Synthesis of (-)-Cylindrocyclophanes A and F Taking advantage of the reversible nature of the olefin cross metathesis reaction. Efficient synthesis of α-aryl esters by room temperature palladium-catalyzed coupling of aryl halides with ester enolates. Palladium- and nickel-catalyzed cross-couplings of unsaturated halides carrying relatively acidic protons with organozinc reagents.
Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. A revised modular approach to (–)-Trans-Δ8-THC and derivatives through late Suzuki-Miyaura cross-coupling reactions. Synthesis of muscothiazoles A and B: Critical role of methyl group substitution in RCM-based syntheses of macrocycles.
Synthesis of novel estrogen receptor antagonists using metal-catalyzed coupling reactions and characterization of their biological activity.