I am also indebted to all the people who populated the Lambert lab during my tenure there. To the newest members of the lab, don't forget to have fun while you work hard.
ABSTRACT
480 Figure A8.2.2.1 Proposed catalytic cycle for the α-arylation of cyclic ketones using in situ generated enolates. 488 Figure A8.2.2.2 Proposed catalytic cycle for the α-arylation of cyclic ketones using in situ generated enolates.

LIST OF SCHEMES
481 Scheme A8.1.1.2 Improved catalyst systems for α-arylation by Hartwig (A) and Buchwald (B) 482 Scheme A8.1.2.1 Current state of the art in catalytic asymmetric α-arylation to form α-.
LIST OF TABLES CHAPTER 1
LIST OF ABBREVIATIONS
INTRODUCTION
- Palladium catalyzed allylic alkylation
- Palladium-catalyzed allylic alkylation of cyclobutanones
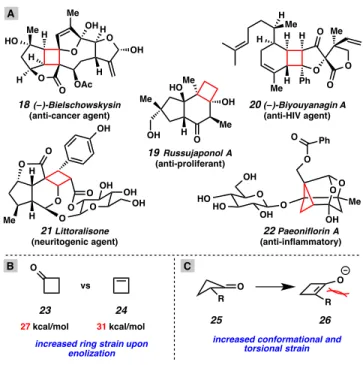
Although methods exist for the alkylation of a variety of enolate types (e.g., ester, ketone, amide, etc.) with a variety of alkylating agents, catalytic enantioselective variants of these transformations are relatively rare.5 Of the available catalytic asymmetric methods, there have been few examples of general techniques for the asymmetric alkylation of carbocyclic systems, and even fewer that have the capacity to. In 2003, we began a program for the catalytic enantioselective synthesis of all-carbon quaternary stereocenters by allylic alkylation of prochiral cyclic ketone enolates.
PREPARATION OF CYCLOBUTANONE β -KETOESTER SUBSTRATES AND REACTION OPTIMIZATION
- Cyclobutanone β-ketoester substrate synthesis
- Optimization of cyclobutanone allylic alkylation
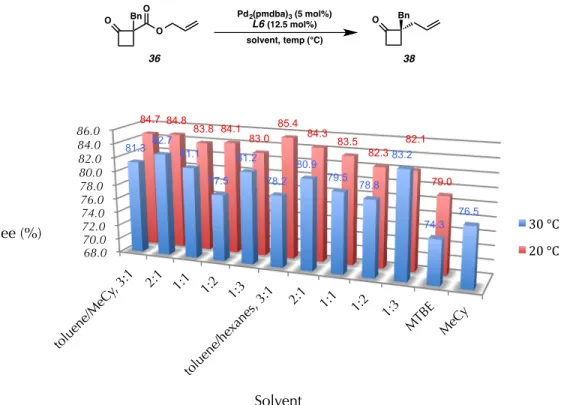
To fully explore solvent effects on reaction selectivity, we used the reaction automation system at the Caltech Center for Catalysis and Chemical Synthesis. Finally, these studies revealed that, in most cases, lowering the temperature at which the reaction was carried out resulted in an increase in selectivity.
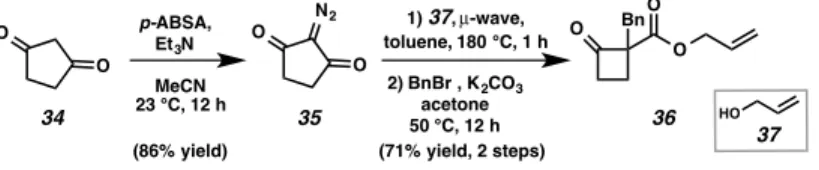
EXPLORATION OF THE REACTION SCOPE
- Reaction scope with respect to enolate α -substitution
- Reaction scope with respect to allyl coupling partner substitution
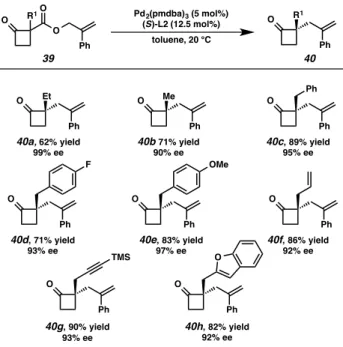
We were pleased to find that α-alkyl substituents were well tolerated with enantiomeric excess up to 99% (Figure a-40b). In addition to alkyl- and benzyl-substituents, allyl-protected 2-carboxyallyl cyclobutanones, TMS, and heteroaryl-substituted cyclobutanones proved to be acceptable substrates in the asymmetric allylic alkylation reaction, providing cyclobutanones 40f-40h with a high excess of production.
DERIVATIZATION OF REACTION PRODUCTS
CONCLUDING REMARKS
EXPERIMENTAL SECTION .1 Materials and Methods .1 Materials and Methods
- Representative procedure for the preparation of 2- oxocyclobutanecarboxylates
- Representative procedure for the alkylation of 2-H-2- oxocyclobutanecarboxylates
The mixture was heated to reflux until complete consumption of the starting material was indicated by TLC analysis (alkylation reaction times typically ranged from 12 to 24 hours). After completion, the mixture was cooled to 25 °C, the solids were removed by filtration through filter paper and the mixture was concentrated in vacuo.
2-Phenylallyl 1-benzyl-2-oxocyclobutanecarboxylate (39c)
2-Phenylallyl 1-(4-fluorobenzyl)-2-oxocyclobutanecarboxylate (39d)
2-Phenylallyl 1-(4-methoxybenzyl)-2-oxocyclobutanecarboxylate (39e)
2-Phenylallyl 1-allyl-2-oxocyclobutanecarboxylate (39f)
2-Phenylallyl 1-(benzofuran-2-ylmethyl)-2-oxocyclobutanecarboxylate (39h)
2-Methylallyl 1-benzyl-2-oxocyclobutanecarboxylate (41b)
2-Chloroallyl 1-benzyl-2-oxocyclobutanecarboxylate (41c)
4-(Benzyloxy)-2-methylenebutyl 1-benzyl-2-oxocyclobutanecarboxylate (41d)
2-(3-Methoxyphenyl)allyl 1-benzyl-2-oxocyclobutanecarboxylate (41e)
2-(4-Fluorophenyl)allyl 1-benzyl-2-oxocyclobutanecarboxylate (41f)
Representative procedure for the asymmetric decarboxylative allylic alkylation of 2-oxocyclobutanecarboxylates alkylation of 2-oxocyclobutanecarboxylates
S)-2-Ethyl-2-(2-phenylallyl)cyclobutanone (40a)
R)-2-Benzyl-2-(2-phenylallyl)cyclobutanone (40c)
R)-2-(4-Fluorobenzyl)-2-(2-phenylallyl)cyclobutanone (40d)
R)-2-(4-Methoxybenzyl)-2-(2-phenylallyl)cyclobutanone (40e)
R)-2-Allyl-2-(2-phenylallyl)cyclobutanone (40f)
Cyclobutanone 40h was isolated by flash column chromatography (SiO 2 , hexanes to 10%. EtOAc in hexanes) as a colorless oil. Cyclobutanone 42a was isolated by flash column chromatography (SiO2, hexanes to 5%. Et2O in hexanes) as a colorless oil.
S)-2-Benzyl-2-(2-methylallyl)cyclobutanone (42b)
R)-2-Benzyl-2-(2-chloroallyl)cyclobutanone (42c)
S)-2-Allyl-2-benzylcyclobutanone (38)
R)-2-Benzyl-2-(2-[3-methoxyphenyl]allyl)cyclobutanone (42e)
R)-2-Benzyl-2-(2-(4-fluorophenyl)allyl)cyclobutanone (42f)
Procedures for derivatization of α-quaternary cyclobutanones and determination of absolute stereochemical configuration
The reaction mixture was then acidified to pH 7 with 1N aqueous HCl and extracted with dichloromethane (2 ml x 5). The mixture was allowed to warm to 25°C and stirred for 18 hours, at which point the reaction was determined to be complete by TLC analysis. After stirring for 30 minutes, this mixture was extracted with Et 2 O (5 mL x 3), dried over MgSO 4 and concentrated in vacuo.
The crude product was purified by flash column chromatography (SiO 2 , 1% EtOAc in hexanes to 5% EtOAc in hexanes) to give α-trimethylsilylcyclopentanone as a colorless oil. The mixture was stirred for 24 hours, at which point the reaction was determined to be complete by TLC analysis. The crude oil was purified by flash column chromatography (SiO 2 , hexanes to 1% EtOAc in hexanes) to give cyclopentanone 45 (47 mg, 0.153 mmol, 69% yield over two steps) as a colorless oil.
The crude mixture was washed with H 2 O (5 mL), washed with brine (5 mL), dried over Na 2 SO 4 and concentrated in vacuo. The crude oil was purified by flash column chromatography (SiO 2 , 3% EtOAc in hexanes to EtOAc) to give lactam 46 (16 mg, 0.05 mmol, 22% yield over two steps) as a pale yellow oil. The reaction mixture was heated to 50 °C and stirred for one hour, at which point the reaction was determined to be complete by TLC analysis.
Determination of enantiomeric excess
INTRODUCTION
- Latent enolates: silyl enol ethers
- Latent enolates: β -ketoesters
- Latent enolates: TMSE β-ketoesters
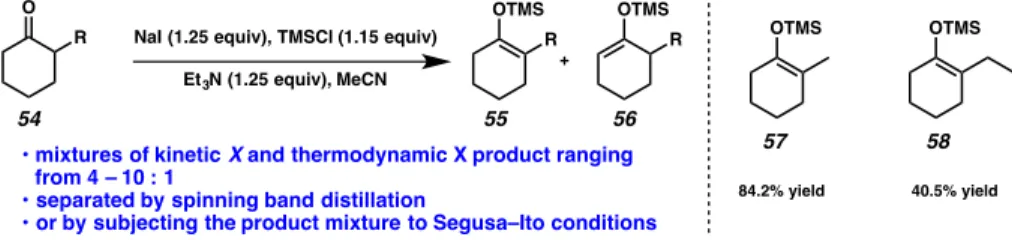
The problem of thermodynamic enolate masking would ideally be solved by the development of enolate precursors that can be easily prepared and, when activated, release the 'thermodynamic' enolate under kinetic control. Allyl-β-ketoesters enjoy relatively straightforward, selective synthesis from simple ketones (i.e., 59) and undergo deprotection upon treatment with a transition metal capable of oxidative addition. Oxidative addition yields a transition metal allyl species, in the present case a palladium π-allyl species 63, and a free carboxylate 62.
In considering new carboxylate-protected enolates, our design criteria required a substrate that could be efficiently synthesized, deprotected under mild conditions, and facilitate convergent coupling of complex fragments in a synthetic environment. These compounds boast similar ease of preparation compared to allyl β-ketoesters, but are not susceptible to transition metal-mediated deprotection. We hypothesized that the use of TMSE β-ketoesters may increase the tolerance width of the functional group on the allylic coupling partner in asymmetric allylic alkylations, relative to allylic β-ketoesters, due to the fact that the allyl fragment is not subject to synthetic conditions of the substrate (Figure 2.1.3.2).
We further reasoned that by eliminating allyl from the reaction mixture, we would overcome the problem of competing reaction pathways in non-allylenolate capture chemistry, and significantly expand the range of reactions in which carboxylate-protected enolates can participate. In this chapter, we describe the preparation and development of this substrate class and its evaluation in the enantioselective palladium-catalyzed allylic alkylation of 6- and 7-membered ketone and lactam scaffolds. Furthermore, we show how the use of these substrates can allow the association of complex fragments carrying functionality that would be incompatible with incorporation into traditional allyl β-.
SYNTHESIS OF AND REACTION OPTIMIZATION WITH TMSE β- KETOESTERS KETOESTERS
- Substrate synthesis
- TMSE-β-ketoester allylic alkylation optimization
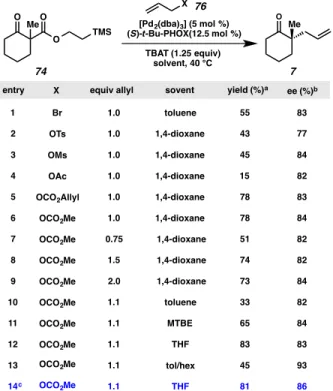
To assess the ability of the substrate to engage in transition metal-mediated catalysis as expected, TMSE-β-ketoester 74 was treated with tetrabutylammonium difluorotriphenylsilicate (TBAT) in THF at room temperature (Scheme 2.2.1.2). This experiment proved the principle that our TMSE-β-ketoesters can indeed undergo mild deprotection and prompted further investigation of the substrate class. With the TMSE β-ketoester 74 in hand, our investigation of this substrate class began in the context of Pd-catalyzed allylic alkylation.
We then explored the range of allyl sources that could be used in the reaction and found that a variety of allyl sources were competent in the chemistry, including allyl sulfonates, allyl acetates, and allyl carbonates (entries 2–5). Allyl methyl carbonate was found to be the most efficient, selective, and cautious allyl source, particularly with respect to the number of allyl equivalents required for optimal reactivity (point 6). TMSE β-ketoester allylic alkylation initial optimization experiments. a) Yield determined by comparison with tridecane internal standard.
The results of these experiments show that the reaction yield is highly variable depending on the solvent used (Table 2.2.2.2), while the selectivity of the reaction remains relatively uniform (Table 2.2.2.3). OCO2Me OCO2Me OCO2Me OCO2Me OCO2Me OCO2Me OCO2Me OCO2Me OCO2Me. The fluctuation in enantioselectivity can be rationalized by a working mechanistic hypothesis for this transformation; in particular, that the enantioselective allylic alkylation proceeds via an inner-sphere pathway,35 and this pathway is enhanced by less polar solvents.
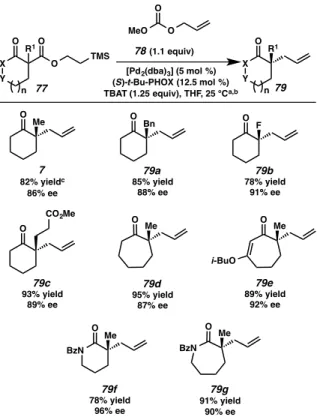
PALLADIUM-CATALYZED ALLYLIC ALYLATION WITH TMSE-β-KETOESTERS .1 Reaction scope with respect to nucleophile
- Reaction scope with respect to electrophile
We were pleased to find that under slightly modified reaction conditions (40 °C), the desired α-functionalized lactam products 79f and 79g were obtained in good to excellent yields and excellent ee. Exploration of functional group and scaffold diversity in the palladium-catalyzed allylic alkylation reaction induced by fluorine in conjunction with nucleophiles. Having observed the extent of the reaction with respect to substitution of the α-nucleophile and the type of scaffold, we next investigated allylic alkylation with respect to substitution at the 2-allyl position.
We were pleased to find that a variety of functional groups could be introduced through the use of differentially substituted allylic carbonates (80, R2 ≠ H, Figure 2.3.2.1). Simple alkyl substitution at the internal allyl position was well tolerated as 2-methylallyl ketone 81a was obtained in 89% yield and 89% ee. Chlorallyl methyl carbonate (80, R2 = Cl) also participated well in the chemistry, providing the corresponding α-quaternary ketone 81b in 72% yield and 96% ee.
Allyl fragments containing neutral and electron-neutral aryl groups also performed well in the reaction, affording the desired allylic alkylation products 81c and 81d , respectively, in excellent yields and ee . Exploration of functional groups and scaffold diversity in the fluorine-induced palladium-catalyzed allylic alkylation reaction with respect to the electrophile.
COUPLING OF TMSE β-KETOESTERS WITH FUNCTIONALLY COMPLEX ELECTROPHILIC PARTNERS ELECTROPHILIC PARTNERS
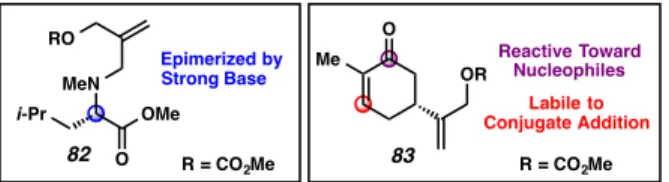
To illustrate this feature of the new chemistry, we synthesized mixed carbonates 82 and 83 as coupling partners for palladium-catalyzed allylic alkylation (Figure 2.4.1). Allyl carbonate 82, derived from leucine, bears an epimerizable stereocenter that racemizes upon strong base treatment.60 Since strong base (i.e., LDA, LHMDS, etc.) is usually required for enolization and acylation in the preparation of β- standard allyl ketoesters, the use of electrophiles with a labile base function has not been possible before.
Alternatively, allylic carbonate 83 , which was synthesized by allylic oxidation of ( S )-carvone, also carries functionality that would be unstable to the conditions required for standard allylic β-ketoester substrate synthesis. In particular, we suggested that attempts to enolate a ketone with an allyl chloro or allyl cyanoformate bearing enone 83 would be hampered by undesirable conjugate addition and enolate chemistry (e.g., Aldol reaction, Michael addition, etc.). In both cases, our new TMSE β-ketoester chemistry allows for the independent preparation and thus physical separation of nucleophilic and electrophilic components up to the fragment coupling stage.
The inherent diastereoselectivity can be completely reversed under catalyst control using ( R )- t -Bu-PHOX ( L9 ), without significant loss in selectivity or reactivity (entry 3).
CONCLUDING REMARKS
We further demonstrated the value of these compounds for joining complex coupling partners, which would be the value of these compounds for joining complex coupling partners that would not be compatible with preparation via standard allyl β-ketoester based allylic alkylation.
EXPERIMENTAL SECTION .1 Materials and Methods .1 Materials and Methods
- General procedure for TMSE β -ketoester substrate synthesis
19F NMR spectra were recorded on a Varian Mercury 300 spectrometer at 282 MHz, and are reported relative to the external standard F3CCO2H (δ –76.53 ppm). IR spectra were obtained using a Perkin Elmer Spectrum BXII spectrometer or Nicolet 6700 FTIR spectrometer using thin films deposited on NaCl plates and reported in frequency of absorption (cm-1) . Analytical HPLC was performed with an Agilent 1100 Series HPLC using a Chiralpak (AD-H or AS) or Chiralcel (OD-H, OJ-H, or OB-H) columns (4.6 mm x 25 cm) obtained from Daicel Chemical Industries, Ltd.