The last group members I would like to thank are Jade Izaguirre, Jade Williams and Abby Smith. In addition to all my peers at Vanderbilt, I would like to thank my friends and family.
Optimization of an enantioselective synthesis of cyclic carbamates via CO 2 -capture 1
Chiral proton catalysis
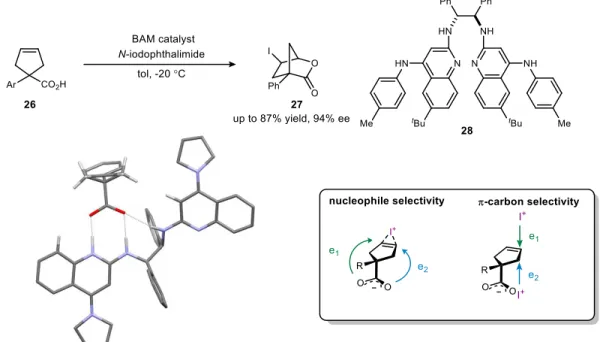
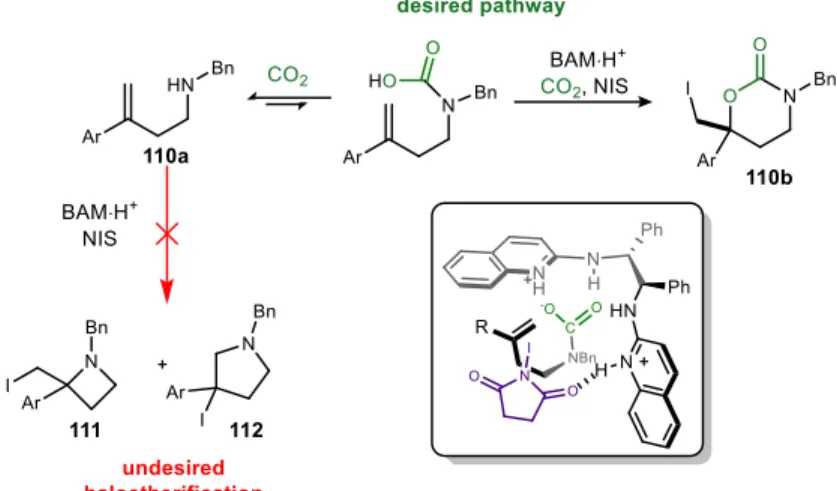
Modifying the quinoline ring substitution allows one to modify the basicity of the chiral ligands. This enabled Brønsted acid activation of the iodine source, which served to activate the olefin.
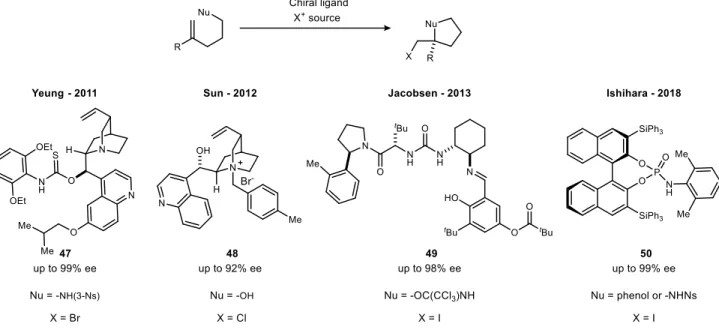
Organocatalytic asymmetric halocyclizations
A wide variety of chiral scaffolds have been prepared for these reaction types, which speaks to the broad utility and applicability of these chiral ligands. This means that the high enantioselectivity of the lactone must be a consequence of high selectivity in the ring-closing step, presumably due to hydrogen bonding to the catalyst.

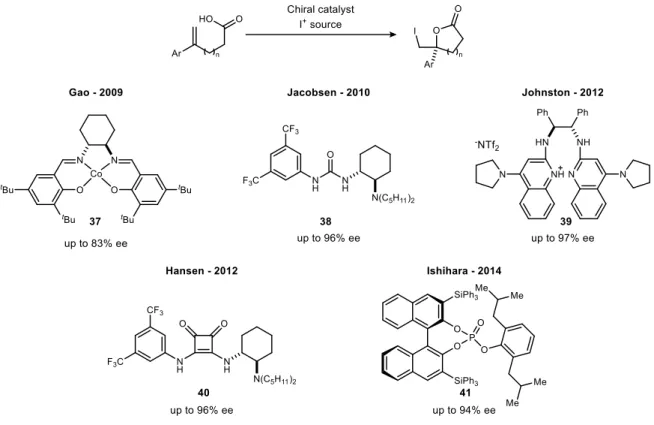
The application of carbon dioxide as a reagent in enantioselective synthesis
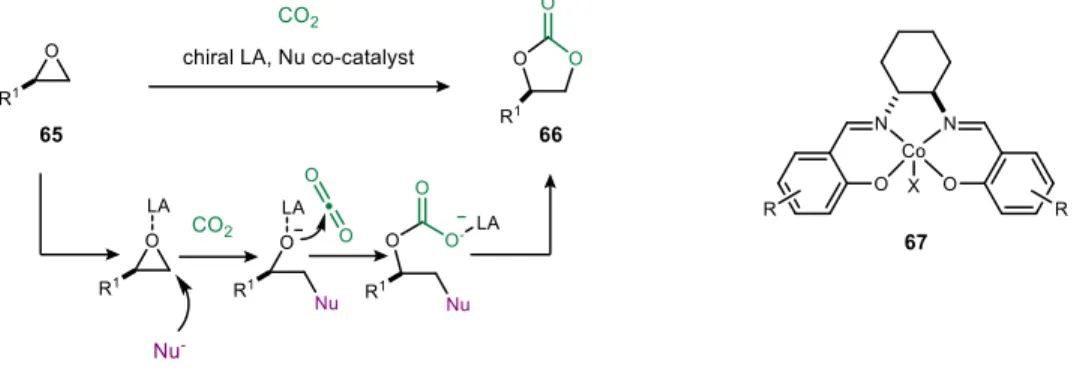
One of the first examples of CO2 capture in enantioselective catalysis comes from the Ihara laboratory for the preparation of cyclic carbonates.52 As shown in Figure 18, the CO2 byproduct from the decarboxylation step is recycled in the cyclization sequence. One of the most common approaches to the cooperative activation of CO2 is the activation of epoxides and aziridine rings to prepare cyclic carbonates and polycarbonates.
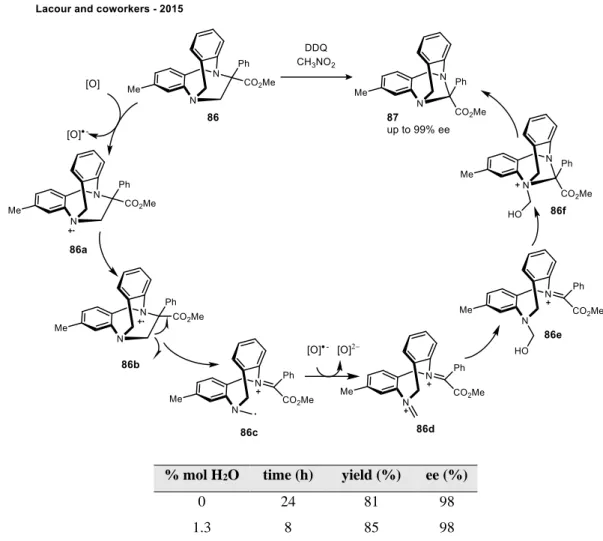
The effect of water as an additive in organic reactions
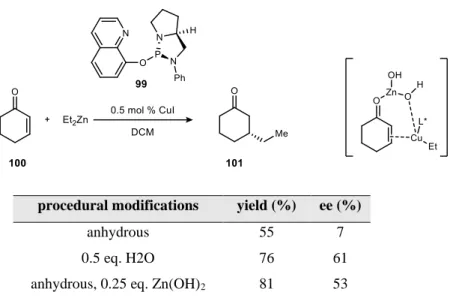
Nevertheless, this is still an interesting example of water as an additive serving as an internal quench agent. This is an example of water acting as a coactivator, making zinc a more effective Lewis acid.
Development of enantioselective carbamation reaction via CO 2 capture
- Proposal for an enantioselective CO 2 capture halocyclization reaction
- Initial reaction optimization
- Observed rate acceleration in wet toluene
- Mechanistic proposal based on DOSY NMR experiments
- Modification of ligand purification to improve catalyst performance
- Analysis of scope and limitations
- Derivation of cyclic carbamates and target synthesis
- Conclusions
It was not understood why the amine substrate became less soluble during the course of the reaction. Water was observed to inhibit the formation of the linear carbonic acid intermediate, which was required for reactivity.

Future directions
Using other nucleophiles that are basic will be a challenge, but a carbonyl can be a sufficient H-bond acceptor for activation. Carbon disulfide would be an interesting area of study because S-nucleophiles have not been studied in BAM catalysis.
Introduction
- Organocatalyzed additions to nitroalkenes
- Organocatalyzed nucleophilic azidation reactions
- Aryl triflamides as achiral modifiers
Nitroalkanes are strong carboxylic acids, due to the strong electron withdrawing effect of the nitro group. The Xiao group used the same thiourea ligand to effect the addition of oximes to nitroalkenes as a method to prepare enantioenriched aminoalcohols.114 Both the oxime and the nitroalkane can be selectively deprotected, allowing selective functionalization of the . Importantly, overprotonation of the ligand weakens (or destroys) basicity and reduces catalyst activity and selectivity.21.
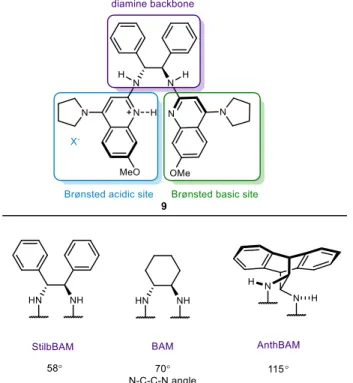
Aryl triflamide is tightly bound to the ligand, greatly reducing the size of the ligand pocket. Changing the substitution pattern greatly affected the size of the catalyst pocket in the solid state. Although the aryl bis(triflamide) residues bind differently to the BAM ligand, both structures show bidentate coordination of the achiral modifier to the chiral ligand.
This second example is an excellent illustration of the achiral modifier effect we hope to design. One side of the ligand pocket is obstructed by an aryl ring, while the other side is open for substrate coordination. This will greatly limit the rotation of the substrate in the catalyst pocket and thus affect the face selectivity.

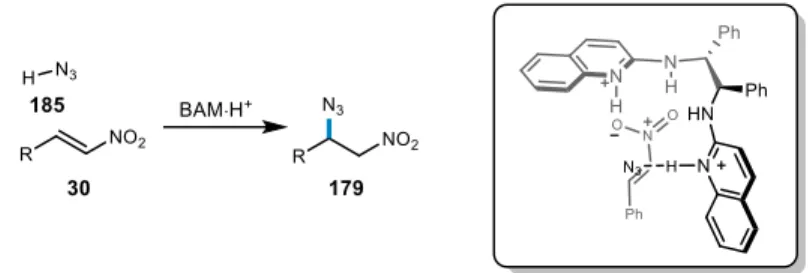
Development of azide addition to nitroalkenes
- Proposal for enantioselective azide addition to nitroalkenes
- Initial reaction optimization
- Application of aryl triflamides as an achiral modifier in BAM catalysis
- Mechanistic proposal for enantioselection
- Analysis of scope in Michael additions
- Derivation of β-azido nitroalkanes
- Synthetic routes to N-aryl triflamides
- Applications of aryl triflamides in other organocatalyzed reactions
- Conclusions
Coordination of the aryl triflamide to the BAM ligand had a significant effect on selectivity, so enhancing this effect may improve selectivity by creating a 'tighter' pocket. Aryl triflamide 194 was found to be the most effective acid for the preparation of the BAM catalyst. It is assumed that two triflamide residues are required because there is a bidentate coordination of the aryltriflamide residue to the amidine moiety of the BAM ligand.
One of the first areas we hoped to investigate was the acidity of the aryltriflamamide residue. This indicated that the spatial arrangement of the triflamide residues should be optimized for the BAM ligand. These results do support the hypothesis that the acidity of the aryltriflamamide residue affects reactivity and selectivity.
It is not immediately clear how the aryl triflamide affects the facial selectivity of the azide addition. In the second proposal, the direction of the hydrazoic acid through the aryl triflamide is the source of nucleophilic control. Most of the aryl triflamides provided the lactone in similar fashion, with the exception of 191 .
Future directions
Optimization of this chemistry led to a novel application of aryl triflamides as achiral modifiers in BAM catalysis. In addition to investigating asymmetric azide addition, we have investigated aryl triflamides in other reactions. Because modifications to the acidic Brønsted site of the BAM catalyst affected the enantioselection in the azide addition chemistry, it is assumed that the aryl triflamide design is the optimal design point moving forward.
There are a number of aryl triflamide derivatives that could be investigated in the future (Figure 85). It was hypothesized that 208 is not coordinated to the BAM ligand via bidentate coordination of the triflamides, because the sulfone may participate as a hydrogen bond acceptor. This may promote bidentate coordination between the BAM ligand and bis(triflamide), while also increasing the acidity of the triflamides.
Adding more electron-withdrawing groups can increase the acidity of the aryltriflamide and thus improve enantioselection. This is a complementary approach to prepare a series of aryl triflamides with different acidities without greatly altering the shape of the scaffold. An analogy can be drawn with the addition of azide to nitroalkenes, where the application of aryl triflamides in BAM catalysis significantly improved enantioselection.

Introduction
- Applications and synthesis of chiral ε-lactones
- Mechanistic analysis of PIDA/I 2 as an oxidant system
The prevailing methods to prepare ε-lactones include ring expansions,184 ring-closing metathesis,197 and macrolactonizations.198 Ring expansions, such as the Baeyer-Villager oxidation, are commonly employed because they circumvent entropic barriers to ring-forming reactions.199 While these reactions have been successfully employed in the preparation of 7-membered lactones, these are not enantioselective transformations and will therefore not be rigorously detailed in this section. Attempts have been made to prepare 7-membered rings by lactonization,200,201 although only one enantioselective example has been reported.202 Tang developed an enantioselective bromoesterification of enynes where NBS is activated by a chiral urea catalyst. The development of highly selective methods to prepare ε-lactones can provide access to new chemical space.
The dual oxidant system PIDA/I2, originally studied by Suarez, has been used in a variety of reactions, including oxidative decarboxylations,203 1,5-HAT oxidations,204 and aromatic halogenations.205 It was suggested that the active oxidant under these conditions is acetyl hypoiodite, as shown in Figure 89.206 Several other reagent oxidant combinations with iodine are also believed to generate acetyl hypoiodite in combination, including silver acetate207, mercuric acetate208 and lead tetraacetate.209 Although this species has not been successfully isolated, the combination of PIDA/ I2 has been investigated to investigate this hypothesis. As shown in Figure 90, it is proposed that isolation problems are associated with the decomposition of acetyl hypoiodite to methyl iodide and carbon dioxide, analogous to the Hundsdiecker reaction.211 To investigate this hypothesis, the thermal decomposition of phenyl diacetate (PIDA) was investigated. using gas chromatography.210 It was observed that both methyl iodide and carbon dioxide were produced in 70.4 and 72.5 mol%, respectively, relative to one mole of PIDA. In support of this hypothesis, methyl acetate was detected by GC-MS in another study of acetyl hypoiodite degradation.206 While acetyl hypoiodite has not been characterized, these degradation studies support its formation in PIDA/I2 reactions.
While this oxidant system has been extensively studied, it has mainly been used at elevated temperatures or in light-mediated radical reactions.212 For this reason, it is not clear whether acetyl hypoiodite is the active oxidant in other applications. Hypervalent iodine reagents are reported in a variety of enantioselective alkene functionalizations.213 In many of these reports, chiral iodine(III) catalysts participate in the formation of the halonium ion intermediate.214,215 While acetyl hypoiodite is reported as a more reactive oxidant, there is precedent that PIDA is the active oxidant in alkene functionalizations. The study of acetyl hypoiodite as an oxidant does not necessarily discredit this alternative hypothesis.

Development of an enantioselective lactonization to prepare ε-lactones
- Previous work in the preparation of ε-lactones via iodocyclization
- Continued efforts in the preparation of 7-membered lactones
- Analysis of scope and limitations BAM-catalyzed lactonization reaction to prepare ε-
- Conclusions
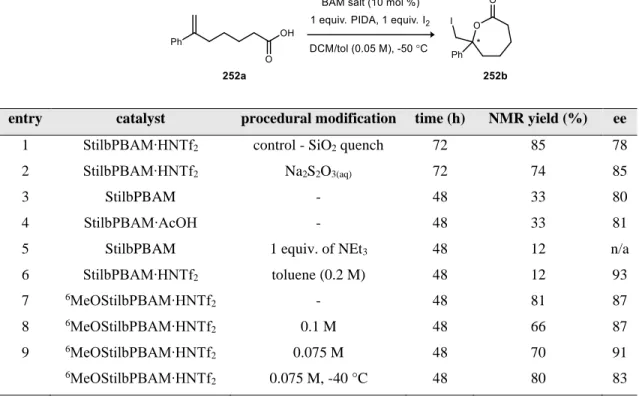
The free base form of the catalyst provided the lactone in a similar ee to the triflimidic acid salt, although the reactivity was significantly reduced. Since coordination of the Brønsted acid to the halogen source is believed to be required for activation, it has been proposed that the active catalytic species is actually a salt of the BAM ligand with an unsaturated carboxylic acid. The acetic acid salt of the ligand BAM was used to test this effect because it is similar in pKa to the substrate.
These results indicate that the protonation state of the catalyst is related to turnover and/or reactivity, but little effect on face selectivity. Because the protonation state of the catalyst affects reactivity, the mode of activation is proposed to be similar to previous BAM-catalyzed cyclizations (Figure 41). The protonated hydrogen bonding site of the ligand binds to the halogenating agent, which provides control of the electrophile (Brønsted acid activation), as shown in Figure 92.
Since the absolute configuration of the lactone is currently undefined, the facial selectivity shown in Figure 92 has not been confirmed. In previous work using silver acetate and iodine as an alternative method to access acetyl hypoiodide, the reactivity was poor.216 As a result, the nature of the active oxidant in this cyclization remains unclear. Optimization of the bisamidine organocatalyst showed that pKa affected reactivity and selectivity, indicating that the basicity of the catalyst can be tailored to the substrate.
Future directions
The reaction was carried out according to the general procedure using cinnamyl methanesulfonate (789 mg, 3.7 mmol) and benzylamine (1.6 mL, 15 mmol). The crude material was separated by silica gel chromatography (SiO2, 10-30% ethyl acetate in hexanes with 0.5% triethylamine) to give a. The reaction mixture was stirred for 2 hours, cooled to room temperature, quenched with saturated aqueous NH 4 Cl and extracted with ethyl acetate.
The reaction vessel was placed under an argon atmosphere, and toluene (16 mL) was measured into the flask. The reaction was cooled to 25 °C, diluted with ethyl acetate and filtered through a celite plug. The reaction mixture was diluted with dichloromethane and transferred to a round bottom flask for evaporation.
After cooling, the reaction mixture was diluted with dichloromethane and transferred to a round bottom flask for evaporation. The crude material was separated by chromatography on silica gel (10-25% ethyl acetate in hexanes) to give impure material. The reaction mixture was diluted with water and transferred to a separatory funnel, where the aqueous layer was extracted with ethyl acetate.
The reaction vessel was placed under an argon atmosphere, toluene (10 mL), tetrahydrofuran (10 mL) and water (5 mL) were measured into the flask. The reaction mixture was concentrated in vacuo and the crude material was dissolved in ethyl acetate.