Therefore, it is very important to study the chemical stability of the QA head groups and the transport of OH- ions at the lowest level of hydration. The degradation mechanisms of QA headgroups under high pH and the transport mechanism of OH- ions in AEM are still active research topics. Therefore, understanding the water structure and ion behavior in these lamellae and confined structures is essential to study the chemical stability of the QA headgroups and the transport of the OH- ion in AEM.
Modeling and simulations at various scales have become a powerful tool to investigate the chemical stability of the QA headgroup-based polymeric matrix of AEM and the transport of OH- ions in AEMFC. Most computational studies on the chemical stability of AEM study through DFT calculations. The effect of C2 substitution on the alkaline stability and degradation reactions of the imidazolium-based head group of AEM were studied.
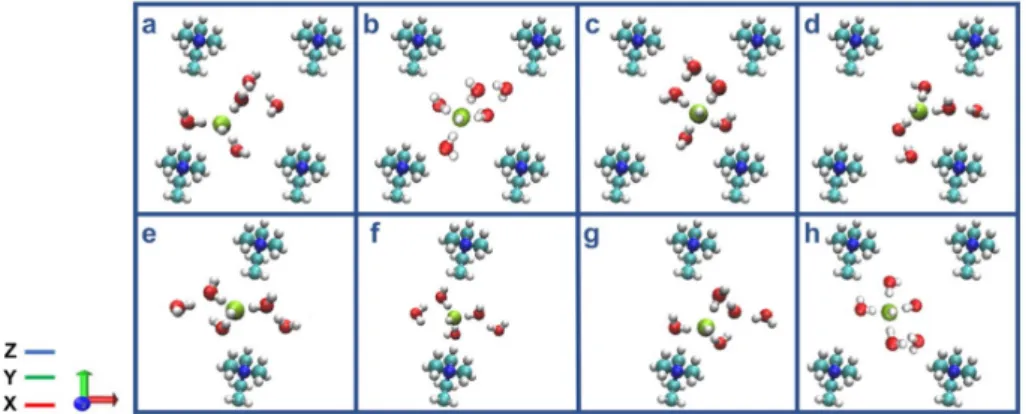
The hydroxide ion is in a fourfold planar structure in the center of the cell, with one water molecule in the second solvation shell. Two water molecules are located at neighboring bottleneck regions. e) The hydroxide ion forms a stable OH˘(H2O)4 complex, in which three water molecules are part of a threefold tetrahedral structure, and one water molecule is in the second solvation shell. A water molecule is located below the hydroxide ion in the bottleneck region. f) The hydroxide ion and five water molecules diffuse through the bottleneck region. g).
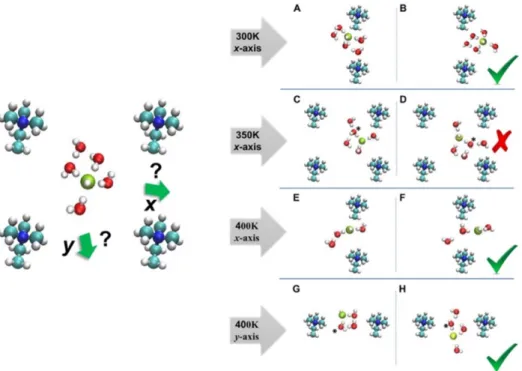
The OH− ion trapped in the center of the AEM was due to the proton rattle event, as shown in Figure 7C,D.

All-Atom Molecular Dynamics (MD) Simulations
The classical all-atom MD simulations for different QA headgroups were performed in the presence of SPC/E water model, OH− ion, with the OPLS force field and LAMMPS software to study the critical relationship between the chemical stability of the QA headgroup in AEMFC environments [166]. The results of classical all-atom MD simulations stated that (i) QA headgroups are unstable at low HL as QAs rapidly decompose, which is consistent with experimental findings, and (ii) high temperature also increases the decomposition of QAs at low HL [166]. The results illustrated that the desired balance between imidazolium group affinity for OH− ion and transport of OH− ion in hydrated imidazolium group grafted PPO chain-based AEM was achieved under critical water saturation conditions, which means 2/8 water molecules were present in the first/second hydration shell [ 167].
In addition, the effect of the imidazolium-based headgroup structure on OH-ion diffusion and chemical stability of AEM was also investigated by classical all-atom MD simulation using Materials Studio, COMPASS II Force field [140]. The results of classical all-atom MD simulations with COMPASS II force field showed that the PPO AEM with 1,2,4,5-tetramethylimidazolium and alkyl spacer chain with six or eight aliphatic carbons at the HL 6 has an excellent balance between chemical stability illustrated. and OH− ion diffusivity for AEM. The diffusion of OH− ion is enhanced by forming a water channel under high water content (Figure 9).
Improved alkaline stability was achieved by replacing the methyl group in the QA headgroup with larger hydrophobic groups, which block the OH ion approaching the nitrogen of QA. Meanwhile, Dubey et al. also classical all-atom MD simulations for 40 monomers of 1 PVB polymer chain with 120–720 water molecules, OPLSAA and SWM4-NDP force fields to study the vehicular diffusion and solvation structure of OH − ions in an AEM [39]. Ethylimidazolium-functionalized poly(arylene ether sulfone) has a high OH-ion conductivity due to well-defined phase separation morphology and chemical stability compared to QA-functionalized.
Classical all-atom models of poly(QPE) based on QA-substituted fluorenyl group, in the presence of water and OH- ion, were simulated via the CVFF force field to study the transport of OH- ions. 172]. Classical all-atom MD models for PEEK (10–40 units), in the presence of OH− ions and water, were simulated via the COMPASS force field and Material Studio software to study the effect of the QA head groups, the polymer, on the transport of hydronium and OH− ions [173]. Classical all-atom MD models for AEM based on trimethyl ammonium PS with OH− and water (TIP5P) were simulated, using DREIDING forcefield and Gromacs software, to study the effect of HL on diffusivity [174] .
At low IEC and water uptake conditions, the OH− ion was poorly hydrated, and the trimethylammonium donated 2–3 coordinating water molecules to balance water distribution within the membrane [174] . The classical all-atom MD simulations mainly enabled us to (i) study nano-phase-segregated water channel and polymer backbone structures of AEM, (ii) calculate the ionic conductivity based on diffusion coefficient using Nernst-Einstein equations, and (iii) the OH− ion diffusion coefficient for Grottus and vehicular transport mechanisms. However, classical all-atom MD is not suitable for the simulation of chemical degradation reactions and transport of OH− ions, as they cannot capture the creation and breaking of chemical bonds.
Coarse-Grained Molecular Dynamics Simulations
Moreover, the PS-based AEM and proton exchange membranes in the presence of water, OH and H+ ions were simulated using the DREIDING force field and LAMMPS software to compare their nanophase-separated structure and transport properties [51]. The better solvation of the QA head groups and OH ions produces less mature hydrogen bonds in the internal structure, especially at lower water content conditions [51]. Several computational works have been carried out to investigate the poly(phenylene oxide), poly(vinyl), poly(arylene ether sulfone), poly(etheretherketone), poly(sulfone), and poly(norbornene) based AEM at the molecular level. .
The rapid development of the QM/MM (ab initio MD) technique can simulate chemical decomposition reactions and OH-ion transport. Electrochemical performance of poly(ether ether ketone) (PEEK)-based AEM CG model. The inset shows the isosurface of W beads at the cut bottom of the simulation field.
As a result, alkyl side chain modifiers achieve nanosegregation of hydrophilic and hydrophobic domains. The results showed that the diffusivity of water and anions increases with the increase of HL and IEC. In addition, the cluster size further increases due to agglomeration with increasing HL or alkyl spacer length [196].
The study of the effect of water content and ionomer architecture on the nanostructure and ionic conductivity of AEM, based on PPO by Lu et al., implies that the ionic conductivity of the AEM is very sensitive to water content but less sensitive to changes in the architecture of the polymer matrix from AEM via CGMD [195]. Therefore, it is suggested that the ionic conductivity of AEM improved through the relationship between polymer chemistry and equilibrium water uptake [195]. Functionalized poly(styrene-b-poly(ethylene-co-butylene)-b-polystyrene-based AEM The DPD simulation for polystyrene-b-poly(ethylene-co-butylene)-b-polystyrene (SEBS), in the presence of water and OH ions, was performed to study the hydrated morphology and microstructure of an alkyl-substituted ionomer [52].
The water distribution did not change, but the backbone structure changed less during functional group selection [200]. The alkyl linker created flexibility in the side chain, leading to an extension of the TMA backbone distance and the uniformity of its distribution. The present studies were essential for the rational design of the effect of polymer architecture, side chain, hydrophobic and ionic interactions and morphology on different polymer-based AEMs.
Continuum Modeling and Simulation
Summary and Outlook
Recent developments in applications of alkaline anion exchange membranes for polymer electrolyte fuel cells.Ind. A mini review of anion exchange membranes for fuel cell applications: stability issues and addressing strategies.Int. Recent trends in the application of PGM-free catalysts at the cathode of anion exchange membrane fuel cells.Curr.
Prediction of the solvation structure and vehicular diffusion of hydroxide ion in an anion exchange membrane using non-reactive molecular dynamics simulation.Chem. Ab initio molecular dynamics study of hydroxide diffusion mechanisms in nanobonded structural mimics of anion exchange membranes.J. Hydroxide ion diffusion in anion exchange membranes at low hydration: Insights from ab initio molecular dynamics.
Molecular dynamics simulation study of a polysulfone-based anion exchange membrane compared to the proton exchange membrane. Effect of water on the stability of quaternary ammonium groups for anion exchange membrane fuel cell applications.Chem. Tunable OH transport and alkaline stability through imidazolium-based groups of poly-(2,6-dimethyl-1,4-phenylene oxide) anion exchange membranes: a molecular dynamics simulation.Ind.
A review on recent developments of anion exchange membranes for fuel cells and redox flow batteries.RSC Adv. The critical relationship between chemical stability of cations and water in anion exchange membrane fuel cells environment.J. Molecular modeling of OH− transport in poly (arylene ether sulfone ketone) s containing quaternized ammonio-substituted fluorenyl groups as anion exchange membranes.J.
Cross-linked poly(vinyl alcohol)/poly(diallyldimethylammonium chloride) as an anion exchange membrane for fuel cell applications.J. Preparation and characterization of crosslinked quaternized poly (vinyl alcohol) membranes for anion exchange membrane fuel cells. Exploring Side Chain Designs for Enhanced Ionic Conductivity of Anion Exchange Membranes Using Mesoscale Simulations.J.
Effect of side chain on the electrochemical performance of poly(ether ether ketone) based anion exchange membrane: A molecular dynamics study. DPD simulations of anion exchange membrane: The effect of an alkyl spacer on the hydrated morphology.
![Figure 1. Schematic illustration of the working principle of AEMFCs. Reprinted with permission from [19]](https://thumb-ap.123doks.com/thumbv2/azdokorg/10963195.0/2.892.252.690.573.944/figure-schematic-illustration-working-principle-aemfcs-reprinted-permission.webp)
![Figure 3. The level of hierarchy from the atomic scale to the system level. Reprinted with permission from [50–53]](https://thumb-ap.123doks.com/thumbv2/azdokorg/10963195.0/5.892.257.774.146.527/figure-level-hierarchy-atomic-scale-level-reprinted-permission.webp)